苯甲酸三氮唑类抗流感病毒化合物及其制备方法和用途与流程
本发明涉及药物化学领域,具体涉及一类苯甲酸三氮唑类抗流感病毒化合物、它们的制备方法以及它们的医药用途。
背景技术:
由病原性微生物流感病毒引起的流行性感冒是一种严重威胁人类生命和健康的呼吸道传染性疾病,其传染性强,分布面广,患者急起高热、全身疼痛、显著乏力。流感病毒可分为不同亚型,如A/California/7/2009(H1N1)、A/Beijing/32/92(H3N2)、A/Vienam/1203/2004(H5N1)、Anhui/1/2013(H7N9)等。流感病毒可以引起非常严重的并发症和死亡,对其预防和控制耗费着大量的社会资源。
鉴于疫苗研发相对滞后,目前临床上使用的特异性抗流感病毒药物主要是M2离子通道阻断剂(如金刚烷胺和金刚乙胺)、神经氨酸酶(NA)抑制剂(如奥司他韦和扎那米韦)和RNA聚合酶抑制剂(如利巴韦林)等。金刚烷胺和金刚乙胺用于流感的预防和治疗已有30多年的历史,但因存在神经毒性和对乙型流感毒无效等缺陷,限制了其临床上的广泛应用。以流感病毒神经氨酸酶抑制剂为代表的抗病毒药物成为迄今抵抗流感最有效的策略。目前临床应用最广泛的药物奥司他韦(Oseltamivir,商品名:达菲)和扎那米韦(Zanamivir,商品名:瑞乐沙)通过与NA催化中心的结合,选择性抑制NA的活性,阻止子代病毒颗粒的释放和扩散,从而有效治疗流感和缓解流感症状。但随着给药的增多,奥司他韦耐药毒株频现,NA的酶活性位点与抑制剂的亲和力降低,极大地限制了对耐药性流感的治疗效果。此外,奥司他韦和扎那米韦由于都含有多个手性中心,合成其光学异构体的步骤繁琐,成本高,使得这些药物价格昂贵。
1999年Wayne J.BrouiLLette等在专利WO99/14191发现了苯甲酸类化合物BANA-206为一个活性较好的苯甲酸类神经氨酸酶抑制剂,其对NA的抑制作用达到纳摩尔级。2006年Li等在Bioorg.Med.Chem.Lett.报道了通过在侧链引入三氮唑类取代基,可得到与扎那米韦活性相近的化合物,该类化合物对细胞培养的流感病毒株具有高选择的抗病毒活性。我们以苯甲酸类化合物作为母核,运用拼合原理和功能团原理,在其3位侧链引入三氮唑类取代基,以期获得高活性的抗流感病毒化合物。
技术实现要素:
本发明公开了一系列苯甲酸三氮唑类抗流感病毒化合物。药理试验结果显示,本发明化合物在体内外对流感病毒及奥司他韦耐药突变株均具有高效的抑制效果。因此,本发明的通式Ⅰ化合物,可用于流感的治疗。
本发明苯甲酸三氮唑类抗流感病毒化合物不含手性中心,合成步骤简便,对多种流感病毒(包括奥司他韦耐药突变株)有效,具有广阔的应用前景。
本发明的化合物通式Ⅰ如下:
其中R1代表:COOH或COO(CH2)n CH3,n=0-6;
R3、R4各自独立地代表:氢或羟甲基,且R3、R4不同时为氢;
R2代表C1-C6的烷基、苯基、苄基、萘基、苯乙烯基或含有1-2个选自O或N的五元或六元杂环基,或被下列任一取代基取代的上述基团:C1-C6的烷基、羟基、硝基、甲氧基、乙氧基、卤素或氨基。
其中R1优选代表COOH、COOCH3或COOCH2CH3。
R2优选代表:
最优选的化合物结构式为:
本发明的部分优选的化合物如下:
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-[5-(2-甲基苯基)-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-1)
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-(-萘基-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-2)
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-(5-仲丁基-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-3)
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-(5-异戊基-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-4)
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-(5-环己基-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-5)
本发明的化合物制备方法如下:
本发明中关键中间体(Ⅱ)3-氨基-4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]苯甲酸乙酯的制备如下:
关键中间体(Ⅲ)3-氨基-4-[2-(羟甲基)-5-氧代吡咯烷酮基]苯甲酸乙酯的制备如下:
本发明开展了该化合物运用于治疗流感病毒感染的研究,在体外通过流感病毒细胞感染模型证明了化合物对甲型流感病毒A/FM/1/47(H1N1)和A/Beijing/32/92(H3N2)具有明显的抑制活性,其EC50值分别为33.6μM和141.7μM,同时该化合物对奥司他韦耐药突变株A/FM/1/47-H275Y(H1N1-H275Y)也表现出显著的抑制作用,EC50值为35.2μM。在体内通过流感病毒小鼠感染模型证明了口服给药本发明化合物对A/FM/1/47(H1N1)感染小鼠有显著的生存保护和体重作用,可明显减轻流感病毒感染症状,延长生存时间。
本发明化合物的药学上可接受的盐具有与化合物同样的药效。所述的药学上可接受的盐优选本发明化合物与下列算加成的盐:盐酸、氢溴酸、甲磺酸、羟基乙磺酸、硫酸、乙酸、三氟乙酸、马来酸、苯磺酸、甲苯磺酸、硝酸、磷酸、硼酸、酒石酸、柠檬酸、琥珀酸、苯甲酸、抗坏血酸或水杨酸等。
本发明还提供了一种治疗流感病毒的药物组合物,其中含有治疗有效量的通式Ⅰ化合物和药学上可接受的载体。所述药物组合物可以是普通片剂或胶囊、缓释片剂或胶囊、控释片剂或胶囊、口服液、注射剂等制剂学上常规的制剂形式。
一般地,本发明的抗流感病毒化合物用于治疗时,人用剂量范围为lmg~1000mg/天。也 可根据剂型的不同和疾病严重程度,使用剂量超出该范围。
下面是本发明部分化合物药效学实验和结果:
一、化合物对流感病毒A/FM/1/47(H1N1)感染MDCK细胞的抑制活性
MDCK细胞按5×103个/孔浓度接种到96孔板中,置37℃、5%CO2培养箱中培养形成细胞单层。细胞用PBS洗2次,加入100TCID50流感病毒A/FM/1/47(H1N1)感染细胞,每孔100μl,37℃孵育2h后弃去病毒液。分别加入含不同浓度(160、80、40、20μM)本发明化合物和先导化合物BANA-206的维持培养液,实验同时设正常细胞对照组、病毒模型对照组及阳性对照药奥司他韦10μM。MDCK细胞于37℃培养48h,每日观察细胞病变。并用MTT法测定细胞活力,实验共重复3次,计算病毒抑制率。病毒抑制率%=(OD加药组-OD病毒对照组)/(OD正常细胞对照组-OD病毒对照组)×100%
实验结果表明:本发明化合物在160μM和80μM浓度下均表现出明显的抗流感病毒活性,而先导化合物BANA-206在160μM浓度下没有明确的抑制作用,其中化合物Ⅰ-5病毒抑制活性最强,在20μM浓度下仍具有22.6%的抑制率(表1),故优选化合物Ⅰ-5进行下一步抗流感病毒活性研究。
表1化合物对流感病毒A/FM/1/47(H1N1)的抑制活性
二、化合物Ⅰ-5的细胞毒性及其对甲型流感病毒A/FM/1/47(H1N1)、A/Beijing/32/92(H3N2)和奥司他韦耐药突变株A/FM/1/47-H275Y(H1N1-H275Y)感染MDCK细胞病变抑制作用
MDCK细胞按5×103个/孔浓度接种到96孔板中,置37℃、5%CO2培养箱中培养形成细胞单层。将本发明化合物Ⅰ-5用维持培养液配制成不同浓度加入MDCK细胞中,使得孔中药物终浓度为20、40、80、160、320、640μM。实验设不加药的正常细胞对照组,将细胞置于37℃、5%CO2培养箱中培养。培养48h后,用MTT法测定细胞活力,确定最大无毒浓度,实验重复 3次,并计算化合物对MDCK细胞的半数毒性浓度(CC50)。
表2化合物Ⅰ-5对MDCK细胞的细胞毒性
根据化合物Ⅰ-5在MDCK细胞上毒性实验结果(表2),实验设置正常细胞对照组、病毒模型组、阳性对照奥司他韦10μM和化合物Ⅰ-5组(设5个不同浓度)。MDCK细胞按5×103个/孔浓度接种到96孔板中,置37℃、5%CO2培养箱中培养形成细胞单层。细胞用PBS洗涤2次,分别加入100TCID50流感病毒A/FM/1/47(H1N1)、A/Beijing/32/92(H3N2)或A/FM/1/47-H275Y(H1N1-H275Y)感染细胞,每孔100μl,37℃孵育2h后弃去病毒液。分别加入含不同浓度Ⅰ-5的维持培养液,MDCK细胞于37℃培养48h,用MTT法测定细胞活力,实验共重复3次,计算病毒抑制率及半数有效浓度(EC50)。病毒抑制率%=(OD加药组-OD病毒对照组)/(OD 正常细胞对照组-OD病毒对照组)×100%
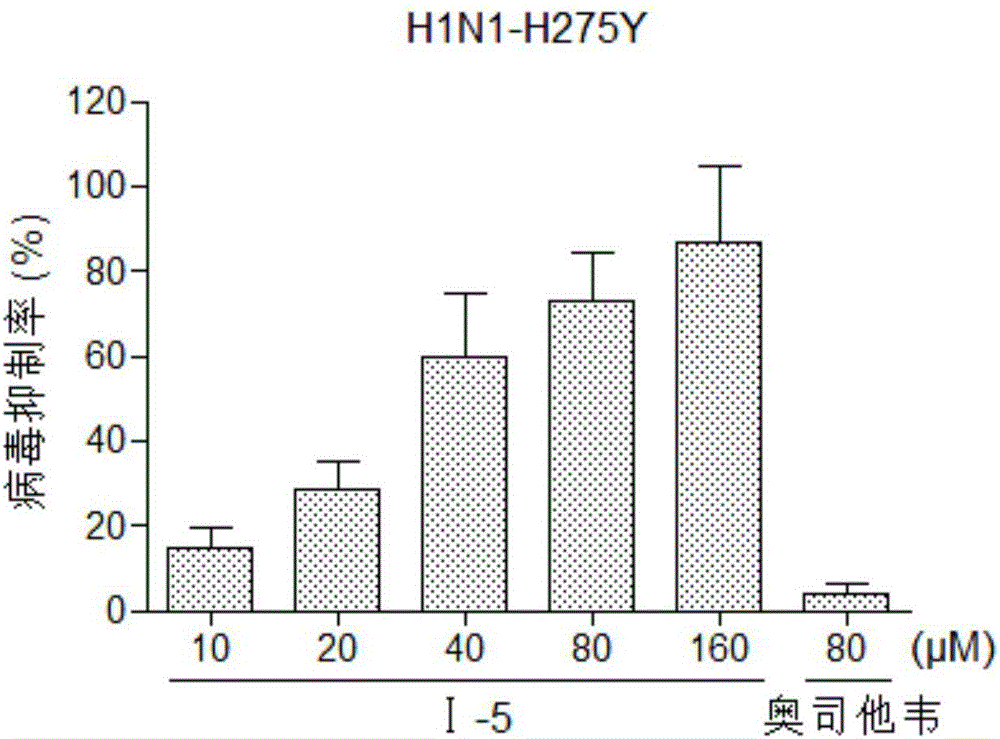
实验结果表明:流感病毒感染MDCK细胞后,导致MDCK细胞产生细胞病变并死亡,致使细胞数目明显减少,细胞活力降低。本发明化合物对流感病毒H1N1和H3N2在MDCK细胞中的复制和增殖具有较强的抑制作用,且呈剂量依赖关系,见图1,对于奥司他韦耐药的突变株H1N1-H275Y也同样有效,见图2,表现出良好的CPE抑制作用,见图3,显著提高了细胞的存活率。本发明化合物浓度为80μM时,对流感病毒H1N1的抑制率达到81.1%,对耐药株H1N1-H275Y的抑制率为72.9%,对流感病毒H3N2的抑制率为42.1%,其EC50值分别为33.6μM,35.2μM和141.7μM,其中对H1N1的抗病毒药效优于H3N2,且有效浓度远低于细胞毒性试验测得的该化合物CC50值(687μM)。
三、化合物Ⅰ-5对甲型流感病毒A/FM/1/47(H1N1)感染小鼠的死亡保护率的影响
实验采用18-20g的ICR小鼠,随机分为4组,设为正常对照组、病毒模型组、阳性对照磷酸奥司他韦组100mg/kg/d、本发明化合物Ⅰ-5组100mg/kg/d。适应性培养7天后开始实验。除正常对照组外,其他各组小鼠用乙醚轻度麻醉,鼻腔内接种相当于含8LD50的流感病毒的鸡胚尿囊液50μl/只,磷酸奥司他韦组和化合物Ⅰ-5组于感染后2h灌胃给药,每日1次,每次0.2ml,连续给药5天。病毒模型组及正常对照组同法给予0.2ml的生理盐水。连续14天观察小鼠生存状况,记录死亡数和死亡时间。
实验结果表明口服给药本发明化合物对流感病毒感染导致的小鼠死亡有较好的保护作用,明显延长流感病毒感染小鼠的存活时间,见图4。给予化合物Ⅰ-5在100mg/kg/d剂量下对 小鼠的死亡保护率为60%,与阳性药奥司他韦效果相当。与病毒模型组相比,该化合物在体内具有明显的抗流感病毒活性,可以显著降低小鼠的死亡率,提高小鼠的死亡保护率,延长存活时间。
四、化合物Ⅰ-5对甲型流感病毒A/FM/1/47(H1N1)感染小鼠的体重保护作用
流感病毒感染小鼠后,引发病毒性肺炎,导致小鼠呼吸不畅、进食减少、体重减轻,所以体重保护是评价药物对病毒在体内增殖的抑制活性的一个重要表观指标。实验结果图5表明,小鼠感染病毒后第5天体重开始下降,并伴随耸毛、食差、消瘦、蜷缩怕冷、低反应性等疾病症状,之后小鼠体重持续降低,病毒模型组下降幅度最大。由于化合物Ⅰ-5和磷酸奥司他韦抑制了病毒在小鼠肺部的增殖,相较于病毒组,给药组小鼠体重下降并不十分明显,在一定程度上表现出对小鼠的体重保护作用。同时,给药组小鼠感染症状相对病毒组得到改善,并且在10天后,体重恢复并增加,部分小鼠的感染症状减少并消失。表明口服给药Ⅰ-5对小鼠体重有一定程度的保护作用,进一步证明了该化合物在体内的抗流感病毒活性。
附图说明
图1是化合物Ⅰ-5对甲型流感病毒A/FM/1/47(H1N1)和A/Beijing/32/92(H3N2)感染MDCK细胞病变的抑制率
图2是化合物Ⅰ-5对奥司他韦耐药突变株A/FM/1/47-H275Y(H1N1-H275Y)感染MDCK细胞病变的抑制率
图3是化合物Ⅰ-5对奥司他韦耐药突变株A/FM/1/47-H275Y(H1N1-H275Y)感染MDCK细胞的保护作用
图4是化合物Ⅰ-5对甲型流感病毒A/FM/1/47(H1N1)感染小鼠的生存保护作用
图5是化合物Ⅰ-5对甲型流感病毒A/FM/1/47(H1N1)感染小鼠的体重保护作用
具体实施方式
实施例1
关键中间体部分的合成
2-[4-(乙氧羰基)苯胺基]丙二酸二乙酯(C)的制备
500mL茄形瓶中加入30g(181.82mmol)对氨基苯甲酸乙酯(A)与20g(84.03mmol)溴代丙二酸二乙酯(B),115℃反应24h后,用乙醚溶解过滤,滤液用10%盐酸洗涤3次,水洗3次,无水硫酸钠干燥。过滤静置析出晶体。抽滤并用少量乙醚洗涤,得23g无色晶体C,产率85.0%。m.p:68~72℃。
MS[M+H]+:324.1;
1H NMR(500MHz,CDCL3),δ:7.9(d,2H,J=8.8Hz,ArH);6.6(d,2H,J=8.8Hz,ArH);5.2(d,1H,J=7.2Hz,CH);4.8(d,1H,J=7.2Hz,NH);4.33~4.37(m,2H,CH2);4.3(m,4H,2CH2);1.35~1.38(t,3H,J=7.2Hz,CH3);1.11~1.14(t,6H,J=7.2Hz,2CH3).
1-[4-(乙氧羰基)苯基]-5,5-二甲酸二乙酯-2-吡咯烷酮(D)的制备
250mL茄形瓶中加入5.60g(17.34mmol)化合物C,50mL无水乙醇,搅拌溶解。量取16mL无水乙醇,切取0.48g(20.81mmol)钠加入其中,制备乙醇钠,将乙醇钠滴加入反应液,反应液变黄。滴加3.69mL(34.68mmol)丙烯酸乙酯溶于10mL无水乙醇的溶液,20℃反应20h后滴加冰乙酸中和旋干。乙酸乙酯、水萃取,无水Na2SO4固体干燥,过滤,柱层析分离得5.58g油状产物,产率85.5%。
MS[M+H]+:378.10;
1H NMR(500MHz,CDCL3),δ:8.01~8.03(d,2H,J=9Hz,ArH),7.38~7.40(d,2H,J=9Hz,ArH),4.33~4.37(m,2H,CH2),4.14~4.19(m,4H,2CH2),2.70~2.73(t,2H,J=7.5Hz,CH2),2.61~2.64(t,2H,J=7.5Hz,CH2),1.35~1.38(t,3H,J=7.2Hz,CH3),1.11~1.14(t,6H,J=7.2Hz,2CH3).
1-[4-(乙氧羰基)-2-硝基苯基]-5,5-二甲酸二乙酯-2-吡咯烷酮(E)的制备
100mL三颈瓶中加入4.66g(12.36mmol)化合物D,23.3mL冰乙酸,34.95mL乙酸酐,搅拌溶解。冰盐浴至-10℃,量取5.83mL(142.76mmol)发烟硝酸,缓慢加入反应液中,反应24h,停止反应。将反应液倒入冰水中,加入Na2CO3固体并搅拌至没有气体生成。再用饱和Na2CO3溶液洗涤3次,再加入无水Na2SO4固体干燥,静置,过滤,旋干得黄色油状液体3.86g,产率74.1%。
MS[M+H]+:424.14.
4-[2,2-(二羟甲基)-5-吡咯烷酮基]-3-硝基苯甲酸乙酯(F)
100mL单颈瓶中加入2.4g(5.69mmol)化合物E。量取24mL无水乙醇、24mL无水THF倒入瓶中,搅拌,冰盐浴至-10℃,分批加入1g(26.40mmol)NaBH4后撤去冰盐浴,室温反应,TLC监测反应完成后,滴加10%HCL中和,旋干溶剂,用乙酸乙酯/水萃取3次,20mL水洗涤2次,加入Na2SO4固体干燥,静置,过滤,柱层析分离得油状物1.06g,产率55.2%。
MS[M+H]+:339.1.
3-氨基-4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]苯甲酸乙酯(Ⅱ)的制备
250mL茄形瓶中加入0.6g(1.78mmol)化合物F。80mL乙醇溶解,油浴50℃。将2.16g(12.46mmol)低亚硫酸钠溶于13mL水中,滴加入上述溶液中,滴加完毕后油浴升温至 75℃,TLC监测反应完成后,冷却至室温,过滤,滤液旋除溶剂,用乙酸乙酯/水萃取,水层反复萃取,收集有机层,无水硫酸钠干燥,静置,过滤,旋除溶剂得黄色油状物0.28g,产率51.1%。
MS[M+H]+:309.2.
1-[4-(乙氧羰基)苯基]-5-甲酸乙酯-2-吡咯烷酮(G)的制备
250mL茄形瓶中加入5.60g(17.34mmol)化合物C,50mL无水乙醇,搅拌溶解。量取16mL无水乙醇,切取0.48g(20.81mmol)钠加入其中,制备乙醇钠,将乙醇钠滴加入反应液,反应液变黄。滴加3.69mL(34.68mmol)丙烯酸乙酯溶于10mL无水乙醇的溶液,加热回流反应20h后滴加冰乙酸中和旋干。乙酸乙酯/水萃取,无水硫酸钠固体干燥,过滤,柱层析(PE:EA=10:1)分离得3.99g油状产物G,产率75.5%。
MS[M+H]+:306.1;
1H NMR(500MHz,CDCL3);δ:8.01~8.04(m,2H,ArH);7.26~7.61(m,2H,ArH);4.75~4.78(m,1H,CH);4.33~4.37(m,2H,CH2);4.16~4.21(m,2H,CH2);2.73~2.77(m,1H,1/2CH2);2.56~2.62(m,1H,1/2CH2);2.46~2.54(m,1H,1/2CH2);2.17~2.23(m,1H,1/2CH2);1.36~1.39(t,3H,J=7.2Hz,CH3);1.19~1.21(t,3H,J=7Hz,CH3).
1-[4-(乙氧羰基)-2-硝基苯基]-5-甲酸乙酯-2-吡咯烷酮(H)
250mL茄形瓶中加入4.66g(15.28mmol)化合物G,23.3mL冰乙酸,34.95mL乙酸酐,搅拌溶解。冰盐浴至-10℃,量取5.825mL(142.76mmol)发烟硝酸,缓慢加入反应液中,反应24h,停止反应。将反应液倒入冰水中,加入Na2CO3固体并搅拌至没有气体生成。再用饱和Na2CO3溶液洗涤3次,再加入无水Na2SO4固体干燥,静置,过滤,旋干得黄色油状液体2.4g,产率45.0%。
MS[M+H]+:351.1;
1H NMR(500MHz,CDCL3);δ:8.64~8.65(s,1H,ArH);8.26~8.28(d,1H,J=10.5Hz,ArH);7.55~7.60(d,1H,J=10Hz,ArH);4.67~4.69(m,1H,CH);4.39~4.44(m,2H,CH2);4.16~4.22(m,2H,CH2);2.60~2.79(m,2H,CH2);2.50~2.57(m,1H,1/2CH2);2.30~2.35(m,1H,1/2CH2);1.36~1.41(t,3H,J=7.2Hz,CH3);1.18~1.23(t,3H,J=7Hz,CH3).
4-[2-(羟甲基)-5-吡咯烷酮基]-3-硝基苯甲酸乙酯(I)
100mL单颈瓶中加入2.4g(6.86mmol)化合物H。量取24mL无水乙醇、24mL无水THF倒入瓶中,搅拌,冰盐浴至-10℃,分批加入0.5g(13.20mmol)NaBH4后撤去冰盐浴,室温反应,TLC监测反应完成后,滴加10%HCL中和,旋干溶剂,用乙酸乙酯/水萃取3次,20mL水洗涤2次,加入Na2SO4固体干燥,静置,过滤,制沙,柱层析(PE︰EA=10︰1)分离得油 状物1.06g,产率50.2%。
MS[M+H]+:309.1;
1H NMR(500MHz,CDCL3),δ:7.57~7.59(d,2H,J=7Hz,ArH);7.12~7.13(d,H,J=8.5Hz,ArH);4.32~4.36(m,2H,CH2);4.08~4.12(m,H,CH);4.16~4.21(m,2H,CH2);3.78(s,1H,OH);3.34~3.47(m,2H,CH2);2.68~2.75(m,1H,1/2CH2);2.55~2.62(m,1H,1/2CH2);2.30~2.38(m,1H,1/2CH2);2.23~2.27(m,1H,1/2CH2);1.35~1.37(t,3H,J=7Hz,CH3).
3-氨基-4-[2-(羟甲基)-5-氧代吡咯烷酮基]苯甲酸乙酯(Ⅲ)
250mL茄形瓶中加入2.01g(6.52mmol)化合物F。80mL乙醇溶解,油浴50℃。将7.94g(45.64mmol)低亚硫酸钠溶于28mL水中,滴加入上述溶液中,滴加完毕后油浴升温至75℃,TLC监测反应完成后,冷却至室温,过滤,滤液旋除溶剂,用乙酸乙酯/水萃取,水层反复萃取,收集有机层,无水硫酸钠干燥,静置,过滤,旋除溶剂得黄色泡沫状物0.7g,产率38.9%。
实施例2
Ⅳ系列中间体的合成
4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]-3-[3-(2-甲基苯甲酰基)硫脲基]苯甲酸乙酯(Ⅳ-1)
将硫氰酸铵0.41g(5.39mmol)和无水丙酮8mL加入25mL三颈瓶中,5min内滴入邻甲基苯甲酰氯0.68g(4.40mmol)溶于15mL丙酮,回流20min,再缓慢滴加3-氨基-4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]苯甲酸乙酯(Ⅱ)0.73g(2.37mmol)溶于15mL丙酮。继续回流12h。减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得到白色固体0.41g,产率35.44%。m.p.141~143℃。
[M+K]+:524.1;
1H NMR(300MHz,DMSO)δ(ppm):12.55(s,1H,NH);11.76(s,1H,NH);9.08(s,1H,ArH);7.85~7.88(m,1H,ArH);7.41~7.54(m,3H,ArH);7.29~7.32(m,2H,ArH);5.31(s,1H,OH);4.89(s,1H,OH);4.31~4.38(q,2H,J=6.9Hz,CH2);3.48~3.54(m,2H,CH2);3.33(s,3H,CH3);3.17~3.22(m,2H,CH2);2.40~2.44(m,2H,CH2);2.21~2.20(m,2H,CH2);1.31~1.35(t,3H,J=7.05Hz,CH3)。
IR(cm-1):3508,3423,3240,1712,1670,1633,1593,1550,1396,1058,856,759.
4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]-3-(3-α-萘甲酰硫脲基)苯甲酸乙酯(Ⅳ-2)
将硫氰酸铵0.41g(5.39mmol)和无水丙酮8mL加入25mL三颈瓶中,5min内滴入α-萘甲酰氯0.90g(4.40mmol)溶于15mL丙酮,回流20min,再缓慢滴加3-氨基-4-[2,2-(二羟甲基)-5- 氧代吡咯烷酮基]苯甲酸乙酯(Ⅱ)0.73g(2.37mmol)溶于15mL丙酮。继续回流12h。减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得白色固体1.02g,收率82.53%。
[M+K]+:560.1;
1H NMR(300MHz,DMSO)δ(ppm):12.60(s,1H,NH);12.03(s,1H,NH);9.10(s,1H,ArH);8.12~8.16(m,2H,ArH);8.03~8.06(m,1H,ArH);7.87~7.90(m,1H,ArH);7.78~7.80(m,1H,ArH);7.54~7.67(m,4H,ArH);5.33(s,1H,OH);4.98(s,1H,OH);4.33~4.37(m,2H,CH2);3.52~3.55(m,2H,CH2);3.20~3.28(m,2H,CH2);2.49~2.50(m,1H,1/2CH2);2.45~2.49(m,2H,CH2);1.32~1.36(t,3H,J=7.05Hz,CH3);1.02~1.07(m,1H,1/2CH2).
IR(cm-1):3417,3236,2977,2360,1714,1523,1496,1396,1292,1217,1157,1024,902,783.
4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]-3-(3-仲丁基硫脲基)苯甲酸乙酯(Ⅳ-3)
将硫氰酸铵0.41g(5.39mmol)和无水丙酮8mL加入25mL三颈瓶中,5min内滴入仲丁酰氯0.53g(4.40mmol)溶于15mL丙酮,回流20min,再缓慢滴加3-氨基-4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]苯甲酸乙酯(Ⅱ)0.73g(2.37mmol)溶于15mL丙酮。继续回流12h。减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得白色固体0.88g,收率82.31%。
MS[M+H]+:452.18.
4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]-3-(3-异戊基硫脲基)苯甲酸乙酯(Ⅳ-4)
将硫氰酸铵0.41g(5.39mmol)和无水丙酮8mL加入25mL三颈瓶中,5min内滴入异戊酰氯0.59g(4.40mmol)溶于15mL丙酮,回流20min,再缓慢滴加3-氨基-4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]苯甲酸乙酯(Ⅱ)0.73g(2.37mmol)溶于15mL丙酮。继续回流12h。减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得白色固体0.97g,收率88.10%。
MS[M+H]+:466.20.
4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]-3-(3-环己基甲酰硫脲基)苯甲酸乙酯(Ⅳ-5)
将硫氰酸铵0.41g(5.39mmol)和无水丙酮8mL加入25mL三颈瓶中,5min内滴入环己基甲酰氯0.64g(4.40mmol)溶于15mL丙酮,回流20min,再缓慢滴加3-氨基-4-[2,2-(二羟甲基)-5-氧代吡咯烷酮基]苯甲酸乙酯(Ⅱ)0.73g(2.37mmol)溶于15mL丙酮。继续回流12h。减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得白色固体0.59g,收率52.13%。m.p.164~166℃。
HR-MS(ESI):m/z[M+H]+calcdforC23H31N36O6S:478.1901;Found:478.1911。
1H NMR(300MHz,DMSO)δ(ppm):12.45(s,1H,NH);11.43(s,1H,NH);8.93(s,1H,ArH);7.82~7.86(m,1H,ArH);7.46~7.48(d,1H,J=8.28Hz,ArH);5.31(s,1H, OH);4.86(s,1H,OH);4.27~4.34(q,2H,J=7.05Hz,CH2);3.07~3.11(m,2H,CH2);2.63~2.68(m,2H,CH2);2.35~2.47(m,2H,CH2);2.14~2.29(m,2H,CH2);1.90~1.93(m,2H,CH2);1.74~1.76(m,2H,CH2);1.62~1.67(m,1H,CH);1.41~1.51(m,2H,CH2);1.30~1.38(m,2H,CH2);1.17~1.26(m,2H,CH2)1,.27~1.32(t,3H,J=7.14Hz,CH3).
IR(cm-1):3419,2933,1718,1683,1583,1535,1290,1159,1018,941,769.
实施例3
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-[5-(2-甲基苯基)-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-1)的制备
将化合物(Ⅳ-1)0.25g(0.51mmol),1滴乙酸酐和0.1ml水合肼在10ml无水乙醇中混合,N2保护下,回流6小时,减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得淡黄色油状物0.15g,收率62.7%。将此油状物用10ml甲醇溶解,加入2N NaOH溶液1ml,室温搅拌,TLC监测至原料点消失,旋除甲醇,加入10%HCl调PH=2~3,有白色固体析出,抽滤烘干得白色固体40mg,产率29.0%。m.p.224~226℃
[M+H]+:438.2;
1H NMR(300MHz,DMSO)δ(ppm):13.23(s,1H,COOH);9.03(s,1H,NH);8.91(s,1H,NH);7.65~7.68(m,1H,ArH);7.40~7.45(m,1H,ArH);7.28~7.34(m,1H,ArH);7.19~7.23(m,1H,ArH);5.18(s,1H,OH);4.11~4.12(m,1H,CH);4.04~4.12(m,2H,CH2);1.87(m,1H,CH).
IR(cm-1):3409,3244,2925,2366,1683,1650,1637,1600,1558,1508,1456,1436,1031,985.
实施例4
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-(-萘基-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-2)的制备
将化合物(Ⅳ-2)0.20g(0.38mmol),1滴乙酸酐和0.1ml水合肼在10ml无水乙醇中混合,N2保护下,回流6小时,减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得黄色油状物0.19g,收率47.80%。将此泡沫状物用10ml甲醇溶解,加入2N NaOH溶液1ml,室温反应,TLC监测至原料点消失,旋除甲醇,加入10%HCl调PH=2~3,有灰白色固体析出,抽滤烘干得灰白色固体39mg,收率21.7%。m.p.248~250℃。
[M-H]-:472.1;
1H NMR(300MHz,DMSO)δ(ppm):13.23(s,1H,COOH);8.91(s,1H,NH);8.07(s,1H,NH);7.91~7.97(m,1H,ArH);7.58~7.64(m,1H,ArH);7.45~7.47(m,1H, ArH);7.19~7.23(m,1H,ArH);5.18(s,1H,OH);4.11~4.12(m,1H,CH);4.04~4.12(m,2H,CH2);1.87(m,1H,CH).
IR(cm-1):3411,2925,1662,1654,1635,1596,1558,1508,1434,1398,1211,1026,808,773.
实施例5
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-(5-仲丁基-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-3)的制备
将化合物(Ⅳ-3)0.25g(0.55mmol),1滴乙酸酐和0.1ml水合肼在10ml无水乙醇中混合,N2保护下,回流6小时,减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得黄色油状物0.20g,收率84.37%。将此泡沫状物用10ml甲醇溶解,加入2N NaOH溶液1ml,室温反应,TLC监测至原料点消失,旋除甲醇,加入10%HCl调PH=2~3,有白色固体析出,抽滤烘干得白色固体34mg,产率24.27%,mp:155~156℃。
MS[M+H]+:404.1;
1H NMR(500MHz,CDCl3);δ(ppm):12.85(s,1H,NH);8.94(s,1H,NH);8.67(s,1H,ArH);7.37~7.40(d,1H,J=8Hz,ArH);7.26~7.28(d,1H,J=8Hz,ArH);3.32~3.36(m,3H,CH2);3.08~3.11(m,1H,1/2CH2);2.49~2.50(m,1H,CH);2.36~2.47(m,2H,CH2);2.24~2.30(m,1H,1/2CH2);2.15~2.20(m,1H,1/2CH2);1.65~1.70(m,1H,1/2CH2);1.54~1.59(m,1H,1/2CH2);1.21~1.23(m,3H,CH3);0.81~0.84(t,3H,J=7Hz,CH3);
IR(cm-1):3543,3414,1681,1635,1616,1556,1384,1120,623,484.
实施例6
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-(5-异戊基-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-4)的制备
将化合物(Ⅳ-4)0.25g(0.54mmol),1滴乙酸酐和0.1ml水合肼在10ml无水乙醇中混合,N2保护下,回流6小时,减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得黄色油状物0.22g,收率91.96%。将此泡沫状物用10ml甲醇溶解,加入2N NaOH溶液1ml,室温反应,TLC监测至原料点消失,旋除甲醇,加入10%HCl调PH=2~3,有白色固体析出,抽滤烘干得白色固体34mg,产率24.27%,mp:155~156℃。
MS[M+H]+:418.2;
1H NMR(500MHz,CDCl3);δ(ppm):12.98(s,1H,NH);8.94(s,1H,NH);8.68(s,1H,ArH);7.37~7.38(m,1H,ArH);7.26~7.28(d,1H,J=8Hz,ArH);5.86(s,1H,OH);5.20(s,1H,OH); 3.32~3.36(m,3H,CH2);3.08~3.11(m,1H,1/2CH2);2.50~2.63(m,1H,1/2CH2);2.36~2.47(m,2H,CH2);2.24~2.30(m,1H,1/2CH2);2.14~2.20(m,1H,1/2CH2);1.61~1.68(m,4H,2CH2);0.77~0.80(t,6H,J=7Hz,2CH3);
IR(cm-1):3416,1647,1618,1552,1452,1384,1257,1072,626.
实施例7
4-[2,2-(二羟甲基)-5-氧代吡咯烷基]-3-(5-环己基-4H-1,2,4-三氮唑基-3-氨基)苯甲酸(Ⅰ-5)的制备
将化合物(Ⅳ-5)0.41g(0.86mmol),1滴乙酸酐和0.17ml水合肼在10ml无水乙醇中混合,N2保护下,回流6小时,减压蒸去溶酶,残留物用柱层析分离(CH2CL︰CH3OH=50︰1),得白色泡沫状物0.23g,收率58.13%。将此泡沫状物用10ml甲醇溶解,加入2N NaOH溶液1ml,室温反应,TLC监测至原料点消失,旋除甲醇,加入10%HCl调PH=2~3,有黄色固体析出,抽滤烘干得黄色固体80mg,产率36%。m.p.228~230℃。
HR-MS(ESI):m/z[M+H]+calcdforC21H27N5O5:430.2085;Found:430.2079.
1H NMR(500MHz,DMSO)δ(ppm):12.92(s,1H,COOH);8.92(s,1H,NH);8.65(s,1H,NH);7.70~7.72(m,1H,ArH);7.34~7.38(m,1H,ArH);7.25~7.27(m,1H,ArH);5.85(s,1H,OH);5.20(s,1H,OH);3.07~3.11(m,2H,CH2);2.63~2.68(m,2H,CH2);2.35~2.47(m,2H,CH2);2.14~2.29(m,2H,CH2);1.90~1.93(m,2H,CH2);1.74~1.76(m,2H,CH2);1.62~1.67(m,1H,CH);1.41~1.51(m,2H,CH2);1.30~1.38(m,2H,CH2);1.17~1.26(m,2H,CH2).
IR(cm-1):3320,2854,1750,1654,1628,1586,1458,1421,1378,1291,1046,860,753。
- 还没有人留言评论。精彩留言会获得点赞!