使用链置换聚合酶的固相靶核酸捕获和复制的制作方法
本公开涉及定性和/或定量检测生物样品中的多种核酸靶序列的核酸测试所需的方法。所述核酸靶序列通常以非常小的量存在于无关核酸序列的大背景中,并且可靠的检测需要大量和高度特异性扩增。本公开的方法提供在使用链置换聚合酶以在温和条件下实现链分离的过程中将通用引发序列与靶序列连接。这些方法适用于具有相对简单装置的自动化。
背景技术:
用于核酸分析的大多数方法提供有限的多路复用能力-例如,由于在高浓度下复杂引物混合物之间的相互作用,标准多重聚合酶链式反应(pcr)的方法限于少数不同的靶标。虽然多重pcr通常用于从同一样品中扩增多种靶标,但由于非特异性扩增、优先靶扩增和引物二聚体假象的可能性,需要进行仔细优化。引物二聚体假象被有效地扩增,这不仅降低了扩增的特异性,而且消耗pcr试剂并降低扩增子产量(elnifro等人,clinmicrobiolrev.13:559,2000;shum和paul,analbiochem388:266,2009)。例如,在基因分型分析中,需要用选定的引物对汇集物(pool)进行6个单独的pcr反应以扩增31种不同的基因座(pastinen等人,genomeres.10:1031,2000)。
已经改进了多重pcr反应的一致性,并且通过使用通用引物进行扩增使引物二聚体的有害作用最小化。
美国专利第5,422,252号、第5,470,723号和第5,648,211号已经公开了这样的方法,其中可以用靶特异性适配子引物(adapterprimer)将通用序列附加到多种靶序列,使得在被称为链置换扩增(sda)的等温方法中可以用单对引物进行扩增。在比使用通用扩增引物时的浓度大幅降低的浓度下,通过使用多对靶特异性适配子引物而使通过其产生的引物二聚体假象最小化。
美国专利第5,882,856号和第6,207,372号已经公开了通用引物在多重扩增中的用途,其在不必针对各靶标进行优化的标准条件下避免单个聚合酶链式反应(pcr)中的优先靶扩增。使用通用引物证明了靶序列更均匀的扩增。
然而,使用通用引物倾向于通过靶特异性引物来减少引物二聚体的形成,通过在扩增子的两条链上使用单引物序列而使在pcr的最初阶段不可避免地形成引物二聚体的扩增最小化(brownie等人,nucleicacidsres.25:3235,1997)。在小的引物-二聚体分子的末端之间形成的杂交物形成锅柄结构,导致了其扩增的相对抑制,这是因为锅柄的形成与pcr引物的退火竞争。
美国专利第5,759,773号公开了这样的方法,其中可以在固体支持物上有效地分离由靶核酸、捕获探针和二元检测探针组成的复合物。参见tyagi等人,procnatlacadsci9:5395,1996和hsuih等人,jclinmicrobiol.34,501,1996。在洗涤固体支持物之后,通过用rnaseh酶促消化捕获探针,将靶标和二元检测探针的复合物从固体支持物分离用以进行随后的连接扩增。该方法需要捕获探针是由rna/dna杂交序列组成的,所述序列可被酶有效地消化,因此对于不同类型的靶分子和捕获序列而言缺乏普遍效用。此外,用该方法仅检测到单个靶标。
美国专利第7,955,794号公开了与美国专利第5,759,773号中的方法相似的方法,其中靶分子被捕获在固体支持物上并与二元检测探针复合,随后连接二元探针并用pcr扩增所连接的探针。已显示该方法适合于单次反应中非常大的数目(成千上万)的靶序列的多重扩增。参见fan等人,genomeres.14:878,2004和fan等人,coldspringharbsympquantbiol.68:69,2003。该方法的成功取决于大量的二元检测探针(各自含有通用引物序列)与固体支持物上靶分子的杂交,洗掉过量的检测探针,然后连接并用通用引物扩增。这有效地去除具有通用引物序列的大量未结合的寡核苷酸探针,否则在聚合酶反应中将会产生大量的引物二聚体假象。
虽然上述方法克服了在大量靶序列的可靠和有效扩增中的显著限制,但该方法不适合于在适于临床分析中的常规使用的简单紧凑的装置中进行自动化。它需要使用两种单独类型的固体支持物和95℃的变性步骤以从第二固体支持物中回收产物用于随后扩增。参见http://support.illumina.com/上的“wgdasl_assay_guide_11322443_b”。必须在一种类型的固体支持物上纯化核酸,然后在酶促反应中复制并用配体将其标记。然后,将标记的核酸与寡核苷酸汇集物杂交并在第二类型的固体支持物上被捕获。在洗涤之后,在固体支持物上进行两次酶促反应。在洗涤固体支持物之后,通过在95℃变性从固体支持物洗脱连接的二元探针,用于pcr扩增。
用并入通用引发序列复制的靶核酸可以用一些不同的扩增方法进行扩增,其中可以用通用引物扩增多种靶标。这些方法包括sda的链置换扩增(美国专利第5,422,252号)、转录介导的扩增或tma(美国专利第5,399,491号)和基于核酸序列的扩增或nasba(kievits等人,jvirolmethods35:273,1991)。
这些方法需要在靶核酸的所关注的序列的两端引入引物和/或启动子序列用于随后扩增,并且这需要靶序列的双链拷贝的产生。已使用不同的策略来用扩增所需的具有5’尾序列的引物引发dna拷贝的合成。
sda方法(美国专利第5,422,252号)利用链置换引物以产生最初靶核酸的单链拷贝,其它引物可以与其杂交以在扩增所需的各端产生具有适配子序列的双链拷贝。随后的扩增依赖于限制酶以在双链dna分子中产生缺口,dna靶标通过dna聚合酶从所述双链dna分子进行复制。
tma法的扩增(美国专利第5,399,491号)已用于扩增rna靶序列,并且依赖于rnaseh的活性以在逆转录之后消化最初的rna靶标来产生用于引物杂交的dna拷贝的单链区域以便产生转录所需的双链dna。由于此依赖于rnaseh以产生rna靶标的双链拷贝,因此原始的tma法不适合于dna靶标。美国专利第7,713,697号和第8,183,359号公开了使tma适于dna靶标的方法,所述方法利用置换剂寡核苷酸以提供dna的单链拷贝,其继而可以从不同的引发序列进行拷贝以产生包括tma扩增所需的双链启动子序列的双链dna。
美国专利第8,198,027号公开了用靶特异性捕获寡核苷酸探针在固体支持物上捕获rna靶标的方法以及将逆转录引物寡核苷酸和与第三寡核苷酸连接的靶核酸进行杂交的方法。当逆转录引物在靶rna上延伸并用rnaseh降解rna模板以在延伸产物上产生引发位点之后,第三寡核苷酸与延伸产物杂交并且延伸以产生双链dna产物。该产物含有随后tma扩增所需的引发和启动子序列。除了可以提供共价或非共价连接外,逆转录引物寡核苷酸和第三寡核苷酸可以通过多种聚合物接头连接。接头可以包括核苷酸和/或非核苷酸残基,并且可以为两个寡核苷酸之间的杂交复合物。
转录介导的扩增已经用于有效扩增某些rna靶序列,但该方法需要仔细优化(美国专利第5,399,491号)。rnase并不完全降解与rna模板杂交的rna模板,并且rnase活性为序列依赖性的。仅rna靶序列的一些区域适于tma,并且分析的功效取决于与具体序列一起使用的rnaseh的类型和浓度。这些性质限制了其效用,尤其是高度多路复用分析。
本公开提供了用一种过程多重扩增许多靶核酸的稳健方法,所述过程适于用于常规临床分析的简单紧凑装置的自动化。
概述
本公开涉及在没有将核酸先前纯化时提供高度特异性分离和扩增样品中的多种核酸靶序列的方法。在需要使用单个固体支持基质和温和条件用于核酸链分离以及从固体支持物回收拷贝的过程中,将通用引发序列引入靶标的拷贝中。所述方法可以在相对简单的装置中实施,其中样品、洗涤溶液和试剂通过自动流体技术从一系列储器引入到单个固体支持基质。
在一个实施方案中,提供了从样品中纯化和复制至少一种靶核酸的方法,其包括:
使所述样品和与所述靶核酸中的序列互补的至少一种寡核苷酸捕获探针接触;
在固体支持物上纯化所述靶核酸;
使第二寡核苷酸与不同于捕获探针结合的序列的所述靶核酸的核酸序列杂交以形成包括所述靶核酸、所述寡核苷酸捕获探针和所述第二寡核苷酸的复合物;
使所述复合物与链置换核酸聚合酶接触,从而将包含新合成的链及其模板链的双链体和与模板互补的单链核酸链相分离,从而所述双链体保持连接于所述固体支持物,并且单链拷贝从所述固体支持物被置换出来。
本文公开的一个实施方案提供了从样品中纯化和复制至少一种靶核酸的方法,其包括:
a)使所述样品与具有5’尾的反向引物/捕获探针寡核苷酸接触并孵育以使与在所述靶核酸上的序列杂交的所述反向引物/捕获探针寡核苷酸退火;
b)使所述样品与反向链置换引物寡核苷酸接触并孵育以使与所述靶核酸上的序列杂交的所述反向链置换引物寡核苷酸退火以形成与所述靶核酸结合的反向引物/捕获探针寡核苷酸和反向链置换引物寡核苷酸的复合物;
通过将所述反向引物/捕获探针寡核苷酸上的配体与固体支持物结合而将所述复合物结合于所述固体支持物上,并洗掉除所述复合物外的过量反向引物/捕获探针寡核苷酸和反向链置换引物寡核苷酸和样品组分;
用核酸链置换聚合酶在所述靶核酸上延伸具有5’尾的反向引物/捕获探针寡核苷酸以形成链,并延伸所述靶核酸上的反向链置换引物寡核苷酸,其中来自所述靶核酸的所述反向链置换引物寡核苷酸的延伸置换了延伸的反向引物/捕获探针寡核苷酸以产生单链拷贝,其为靶核酸的拷贝并且其与所述固体支持物连接;
使含有单链拷贝的介质和与所述单链拷贝上的序列杂交的具有5’尾的正向引物以及与所述单链拷贝上的序列杂交的正向链置换引物的混合物接触,并孵育以使正向引物和正向链置换引物退火至单链拷贝以形成复合物并洗掉过量的正向引物和正向链置换引物;
用核酸链置换聚合酶在固定于所述固体支持物上的所述单链拷贝上延伸具有5’尾的正向引物和所述正向链置换引物,其中所述链置换引物的延伸置换了延伸的正向引物链,延伸的正向引物链从单链拷贝中被置换出来;
从含有固体支持物的介质中回收所述延伸的正向引物链,延伸的正向引物链含有所述正向引物的5’尾序列和所述反向引物捕获探针的5’尾的互补物;以及
使用连接的5’尾序列中的一种或两种扩增所述延伸的正向引物链。
在该实施方案中,所述反向引物/捕获探针寡核苷酸和所述反向链置换引物寡核苷酸可以与所述靶核酸同时杂交。
所述反向引物/捕获探针寡核苷酸和所述反向链置换引物寡核苷酸可以与所述靶核酸分别杂交。
在使所述复合物与所述核酸链置换聚合酶接触之前,可以洗涤所述固体支持物以去除任何过量的未与固定于固体支持物上的靶核酸杂交的反向链置换引物寡核苷酸。
本文还公开了从样品中纯化和复制至少一种靶核酸的方法,其包括:
使所述样品与反向引物的混合物接触并孵育以使所述反向引物退火至所述靶核酸中的序列以形成复合物;
用捕获探针/引物将所述复合物结合于固体支持物,其中所述捕获探针/引物与相对于有尾反向引物结合的序列的3’位置的所述靶核酸上的序列杂交,并洗掉除靶序列外的过量的引物和样品组分;
用链置换聚合酶在所述靶核酸上延伸所述反向引物和所述捕获探针/引物,从而从所述固体支持物置换所述靶核酸的单链拷贝并且其中靶核酸/延伸的捕获探针/引物杂交物保留在所述固体支持物上;以及
将与所述单链拷贝上的序列杂交的靶特异性有尾正向引物和2种通用引物的混合物添加至所述靶核酸的单链拷贝并用聚合酶反应扩增所述靶核酸的延伸产物。
在该实施方案中,与所述序列杂交的所述捕获探针/引物包含信使rna的聚腺苷酸化位点。
与所述序列杂交的所述捕获探针/引物包含对于各个靶核酸不同的靶特异性序列。
可以通过参考下述详细描述和附图来实现对本公开的功能性和有利方面的进一步理解。
附图简述
现仅通过示例的方式,并参考附图描述实施方案,其中:
图1为说明本公开内容的实施方案的图,其中利用2个链置换步骤以产生在各拷贝的两端具有并入的通用引发序列的靶序列的单链拷贝并且将其与固体支持物分离用于随后处理。
图2为说明本公开内容的实施方案的图,其中利用1个链置换步骤以产生在各拷贝的一端具有并入的通用引发序列的靶序列的单链拷贝并且将其与固体支持物分离用于后续处理。
图3呈现了以下实验结果,其中本文公开的方法用于检测少量的三种合成的rna分子。
图4呈现了以下实验结果,其中本文公开的方法用于检测少量的hivrna。
图5呈现了以下实验结果,其中本文公开的方法用于检测来自培养的乳腺癌细胞的少量的(33至300pg)人信使rna。
图6呈现了以下实验的结果,其中本文公开的方法用于检测从血液分离的少量的乳腺癌细胞的信使rna。具体而言,四种基因的信号强度使用log10量表。三种样品类型的六个重复:收获的血细胞对照(最不密集的孵化)、具有10个skbr3细胞的收获的血细胞(较密集的孵化)以及具有100个skbr3细胞的收获的血细胞(最密集的孵化)。实心条表示重复的平均值。
图7呈现了以下实验的结果,其中本文公开的方法用于定量来自培养的乳腺癌细胞的少量的(0.05至500pg)人信使rna。
详细描述
将参考下文论述的详细内容描述本公开内容的各个实施方案和方面。下述描述和附图是对本公开内容的说明,并且不应解释为限制本公开内容。在此描述了许多具体细节以提供对本公开内容的各种实施方案的透彻理解。然而,在某些情况下,为了提供本公开内容的实施方案的简洁描述,本发明并未提及公知常识或常规细节。
如本文所用,术语“包含(comprises)”和“包含(comprising)”应被解释为包括性的和开放式的,而不是排他的。具体而言,在本说明书和权利要求中使用时,术语“包含(comprises)”和“包含(comprising)”及其变型意味着包括指定的特征、步骤或组分。这些术语不应被解释为排除了其它特征、步骤或组分的存在。
如本文所用,术语“示例性”意味着“作为实例、例子或说明”,且不应被解释为优于或胜过本文所公开的其它配置。
如本文所用,术语“约”和“大约”意味着涵盖可能存在于数值范围的上限和下限中的变化,如在性质、参数和尺寸中的变化。在一个非限制性实例中,术语“约”和“大约”意味着加或减10%或以下。
除非另有定义,本文所用的所有技术和科学术语旨在具有本领域普通技术人员通常所理解的相同的含义。例如,提及寡核苷酸的5’或3’端或者寡核苷酸上的5’尾是指构成聚合物链的主链的糖部分上的碳原子的编号并且指示链的方向性。本公开的优选实施方案示于图1中。示出的方法的组成如下:
(1)-待纯化和扩增的靶核酸
(2)-反向引物/捕获探针寡核苷酸,其为具有5’尾的反向引物和捕获探针,包括用于固定的配体
(3)-反向链置换引物寡核苷酸
(4)-具有配体结合剂的固体支持物
(5)-固定于所述固体支持物上的单链dna拷贝
(6)-置换的延伸产物/靶杂交物
(7)-具有5’尾的正向引物
(8)-正向链置换引物
(9)-具有正向和反向尾序列的置换的dna拷贝
(10)-保持结合于固体支持物的双链dna
(17)-与反向引物/捕获探针寡核苷酸(2)杂交的靶核酸(1)上的靶特异性序列
(18)-不同于序列(17)并且相对于靶核酸(1)上的序列(17)位于5'方向的靶特异性序列。反向链置换引物寡核苷酸(3)与该序列杂交。
(19)-固定于固体支持物(4)上的单链dna拷贝(5)上的序列
(11)-不同于序列(19)的固定于固体支持物(4)上的单链dna拷贝(5)上的序列
(12)-在单链dna拷贝(5)上具有5’尾的正向引物(7)的延伸,所述单链dna拷贝(5)包括具有5’尾的反向引物寡核苷酸捕获探针(2)的序列的互补物
如下为图1中示出的方法中的步骤:
(a)将针对待纯化和复制的各个靶核酸(1)的2种寡核苷酸(2、3)的混合物添加至样品裂解物中并孵育以使寡核苷酸(2、3)退火至靶核酸(1)以形成复合物。
(b)通过将反向引物(2)上的配体与固体支持物(4)结合在固体支持物(4)上捕获复合物并洗掉除了靶序列外的过量寡核苷酸(2、3)和样品组分。反向引物(2)上配体与固体支持物(4)的结合可以由于反向引物(2)上的配体与先前固定于固体支持物(4)的化学实体(例如,寡核苷酸上的生物素与固体支持物(4)上的链霉亲和素)之间的固有亲和力而同时发生。或者,反向引物(2)可以预先通过共价或非共价手段连接于固体支持物(4)。本领域技术人员会理解这种捕获可以是以任一方式来完成。
(c)用链置换聚合酶在靶核酸(1)上延伸具有5’尾的反向引物(2)以形成链(5),并延伸靶核酸(1)上的反向链置换引物(3)。链置换聚合酶由于其固有活性而实现这一点。该活性是某些核酸聚合酶的性质,其是天然存在的或者是由天然存在的核酸聚合酶工程化的核酸聚合酶中存在的。反向链置换引物(3)的延伸从靶核酸(1)置换了连接于固体支持物(4)的延伸的反向引物(2)(即,链(5),其是靶标(1)的拷贝)。因此,双链延伸产物/靶杂交物(6)从固体支持物(4)被置换出,并在聚合酶反应后被洗掉。双链延伸产物/靶杂交物(6)由靶核酸(1)和靶核酸(1)上的寡核苷酸引物(3)的延伸物组成。两条链之一为靶标(1),且另一条链(5)为由于引物(3)的延伸而产生的靶标(1)的拷贝。
(d)使具有5’尾的正向引物(7)和正向链置换引物(8)退火至固定于固体支持物(4)的靶核酸的单链拷贝(5),并在退火之后洗掉过量的寡核苷酸(7、8)。
(e)用链置换聚合酶在固定于固体支持物(4)的单链dna拷贝(5)上延伸具有5’尾的正向引物(7)和正向链置换引物(8)。链置换引物(8)的延伸从单链dna拷贝(5)中置换了延伸的正向引物链(9)。从浸泡固体支持物(4)的介质中回收延伸的正向引物链(9)(其含有正向引物(7)的5’尾序列和反向引物捕获探针(2)的5’尾的互补物),并且可以使用连接的尾序列5’中的一种或两种对其进行扩增。链(9)显示于图1中,其中5'尾在左侧,并且3'尾在右侧。在右侧的3'尾是引物(2)的5'尾的互补物。
因此,本文公开了从样品中纯化和复制至少一种靶核酸(1)的方法,其包括使样品和与靶核酸(1)中的序列(17)互补的至少一种寡核苷酸捕获探针(2)接触,在固体支持物(4)上纯化靶核酸(1),使第二寡核苷酸(3)与不同于捕获探针(2)结合的序列(17)的靶核酸(1)的核酸序列(18)杂交,以及使包括靶核酸(1)和2种寡核苷酸(2、3)的复合物与链置换核酸聚合酶接触(图1中未示出),从而将包括新合成的链(19)及其模板链(1)的双链体(6)与模板(1)互补的单独新合成的核酸链(5)分离。
捕获探针(2)和第二寡核苷酸(3)可以与靶核酸(1)同时杂交。在由靶核酸(1)、捕获探针(2)和第二寡核苷酸(3)组成的复合物与核酸聚合酶(未示出)接触之前,可以洗涤固体支持物(4)以去除过量的未与固体支持物(4)上捕获的靶核酸(1)杂交的第二寡核苷酸(3)。捕获探针(2)可以在靶核酸序列(1)上延伸,并且第二寡核苷酸(3)可以从捕获探针(2)的延伸产物(5)中置换靶核酸(1),导致固定于固体支持物(4)上的靶核酸(1)的单链复制物(5)。捕获探针(2)可以包括与靶核酸(1)的序列(17)互补的靶特异性序列的5’尾序列。
第二寡核苷酸(3)可以从靶核酸(1)置换捕获探针(2),导致从固体支持物(4)置换包括靶核酸(1)和靶标(1)的复制物(19)的双链杂交物(6)。
在方法中,第三寡核苷酸(7)可以与靶核酸(1)的单链复制物(5)的序列(20)杂交并且可以通过核酸聚合酶延伸,聚合酶未示于图1中。在复合物与核酸聚合酶接触之前,可以洗涤固体支持物(4)以去除过量的未与靶核酸(1)的单链复制物(5)杂交的第三寡核苷酸(7)。
第三寡核苷酸(7)可以包括寡核苷酸的靶特异性部分的5’尾序列,其为靶核酸(1)的单链复制物(5)的序列(20)的互补物。在方法中,第四寡核苷酸(8)可以与不同于第三寡核苷酸(7)结合的序列(20)的靶核酸(1)的单链复制物(5)上的序列(19)杂交,并且该第四寡核苷酸(8)可以通过链置换核酸聚合酶延伸以从固体支持物(4)置换由第三寡核苷酸(7)的延伸产物(21)组成的单链核酸分子(9)。
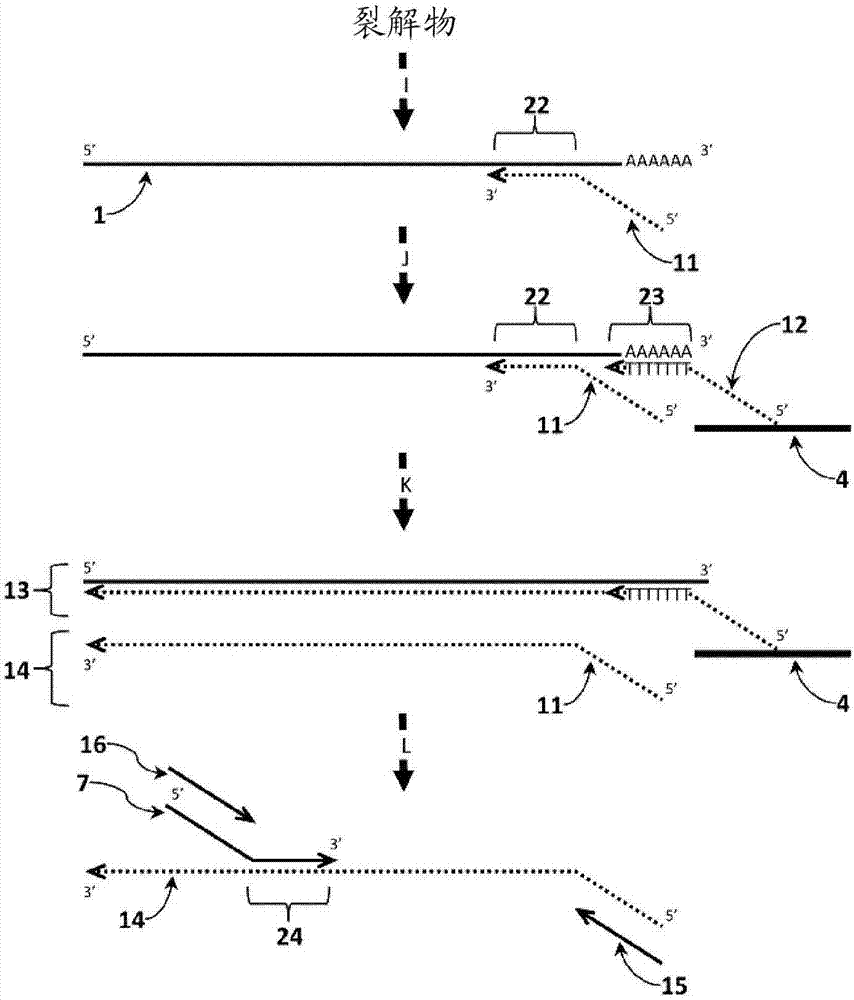
另一实施方案示于图2中。示出的方法的组成如下:
1-靶核酸
4-固体支持物
7-具有5’尾的正向引物
11-具有5’尾的反向引物
12-反向引物/捕获探针,其3’末端与靶核酸序列互补,并且其5’尾端能够与固体支持物基质(4)连接。图2中的引物/捕获探针(12)具有与图1中引物/捕获探针(2)不同的数目,这是因为图2中的引物/捕获探针(12)位于有尾引物(11)的右侧并从固体支持物(4)置换有尾引物(11)的延伸产物。
13-保留在固体支持物(4)上的双链延伸产物/靶杂交物
14-从固体支持物(4)被置换的单链dna拷贝
15-反向通用引物
16-正向通用引物
22-与反向引物(11)杂交的靶核酸(1)上的靶特异性序列
23-不同于序列(22)并且相对于靶核酸(1)上的靶特异性序列(22)位于3'方向上的靶核酸上的序列。该序列(23)可以为样品中多于一种靶序列上所存在的序列,例如,真核生物mrna上的poly-a位点。
24-与具有5’尾的正向引物(7)杂交的从固体支持物(4)被置换的单链dna拷贝(14)上的序列
如下为图2中示出的方法中的步骤:
i.将有尾反向引物(11)的混合物添加至含有靶标(1)的样品中(每种靶标一种引物)并使其退火至靶核酸(1)上的序列(22)。
j.用与相对于由有尾反向引物(11)结合的序列(22)在靶标的3’位置上的序列(23)杂交的捕获探针/引物(12)将复合物捕获在固体支持物(4)上,并洗掉除靶序列(1)外的过量引物(11)和样品组分。
k.用链置换聚合酶(未示出)在靶核酸(1)上延伸反向引物(11)和捕获探针/引物(12),从而从固体支持物(4)置换靶核酸(1)的单链拷贝(14)。靶核酸/捕获探针/引物杂交物(13)保持在固体支持物(4)上。
l.将与单链拷贝(14)上序列(24)杂交的靶特异性有尾正向引物(7)和两种通用引物(15和16)的混合物添加至靶核酸(1)的单链拷贝(14)并用聚合酶反应扩增靶核酸(1)的延伸产物(14)。
图2中的正向靶特异性有尾引物(7)可以以比图2中的正向通用引物(16)显著更低的浓度存在。与正向通用引物(16)相比,图2中的正向有尾引物(7)的靶特异性5'部分也可以具有较低的解链温度。
在本文公开的实施方案中,可以在不使用破坏核酸杂交物的变性条件的情况下于浸泡固体支持物(4)的溶液中回收靶核酸序列(1)和并入的5’尾序列的所置换的单或双链复制物。
另外,可以在不使用化学切割方法的情况下于浸泡固体支持物(4)的溶液中回收靶核酸序列和并入的5’尾序列的所置换的单或双链复制物。
正向和反向靶特异性引物(图1中的7和2以及图2中的7和11)的5’尾序列可以提供用于随后扩增的引发位点。可以用聚合酶链式反应实现该扩增。或者,可以用链置换扩增反应实现该扩增。
尾序列可以提供用于随后扩增的启动子位点,并且可以通过转录介导的扩增或nasba实现该扩增。
可以通过多种方法分析扩增产物,所述方法包括通过在固定于固体支持物(如微阵列)或固定于不同珠粒的多种不同探针上进行杂交,通过实时pcr或通过测定产物的完整序列。对于产物的测序,引物上的5’尾可以含有随后测序反应所需的适配子序列或鉴定特定样品或靶核酸的标签。
固体支持物(4)可以为磁珠、膜或多孔导流(flow-through)芯片。
与序列(23)杂交的捕获探针/引物(12)可以包含信使rna的聚腺苷酸化位点。与序列(23)杂交的捕获探针/引物(12)包含对于各个靶核酸不同的靶特异性序列。
在本发明的方法中,核酸靶序列可以分离自含有去污剂和/或离液剂的粗裂解物,使得可以通过本文公开的方法分离靶核酸而无需核酸的先前纯化。
图2中的方法比图1中的方法简单,但图1中的方法可能比图2中的方法更能抵抗引物二聚体的作用。与图1中的方法相比,图2中的方法对于各个靶序列而言需要更少的寡核苷酸,且涉及更少的步骤。图1的方法中的正向引物在靶核酸的单链拷贝固定于固体支持物时与所述靶核酸的单链拷贝杂交,使得在随后的聚合酶反应之前可以洗掉过量的正向引物。图2的方法中的正向引物在靶核酸的单链拷贝从固体支持物中被置换之后与所述靶核酸的单链拷贝杂交,使得过量的引物不能被洗掉。在这种情况下,引物二聚体可以在不同基因特异性正向引物之间形成。然而,这些引物二聚体假象的形成可通过使引物浓度最小化来限制。此外,引物二聚体的扩增是受限的,这是因为由正向基因特异性引物形成的所有引物二聚体在其5'端具有相同的尾序列且在其3'端具有5'尾序列的互补物,因此它们将形成发夹结构并且不会有效地扩增。
可以制备包含用于实施本公开方法的试剂的试剂盒。
实施例
使用不同类型的样品,包括合成rna(任意随机序列和hiv序列)和来自乳腺癌细胞和全血的总rna,来测试图1中概述的方法。使用来自乳腺癌细胞的总rna来测试图2中概述的本公开的方法。
合成rna(任意随机序列&hiv5’ltr序列)
用合成的rna靶标测试图1中概述的方法。将3种不同的合成rna靶标(各自为约400个碱基)的汇集物掺入含有去污剂的裂解/杂交缓冲液,并允许将所述合成的rna靶标的各自链置换引物和生物素化捕获探针(在5’端含有通用序列)退火。在链霉亲和素磁珠上捕获复合物,并且在逆转录/链置换反应之前,洗掉过量的探针和引物。在逆转录之后,洗掉置换的双链rna:cdna复合物,并且将第二组引物添加至捕获探针的延伸产物以通过第二链置换反应在5’端引入另一通用序列。将上清液添加至含有通用正向引物和生物素化通用反向引物的pcr混合物中,并扩增靶标。在导流寡核苷酸微阵列上杂交生物素化pcr产物(quinn等人,jtranslmed.7:55,2009),随后洗涤,与链霉亲和素-hrp孵育,并进一步洗涤。通过化学发光检测靶标的存在。对于掺入到相同裂解/杂交缓冲液的所有三种合成的rna靶标,检测出低至10飞克(fg)的信号(图3)。
类似地,将具有hiv5’ltr序列的合成的rna(~600个碱基)用于测试图1中概述的方法。在该实验中,将2种不同组的靶向hiv序列的2个不同区域的生物素化捕获探针和引物与合成hivrna杂交并使用前述方法进行处理。对于掺入裂解/杂交方法的合成hivrna靶标,检测出低至100fg的信号(图4)。
skbr3乳腺癌细胞系和通用人类参照总rna
使用来源于哺乳动物skbr3乳腺癌细胞系的总rna来测试用本发明的方法的多种靶标检测的可行性。将包括来源于10种不同细胞系的rna的通用人类参照(uhr)总rna用作对照。将与乳腺癌相关的数种相关基因(erbb2、epcam、krt9、ccnd1)和持家基因actg1选择作为mrna靶标。针对这些靶标中的每一种设计生物素化捕获探针和引物,将样品掺入含有去污剂的裂解/杂交缓冲液,并使用图1概述的方法进行处理。
测试了来自skbr3乳腺癌细胞的不同量的总rna,并对于持家基因actg1和乳腺癌基因的子集,检测出低至33.3pg的信号(图5)。在任何样品中未检测到krt9,这是因为krt9未在该细胞系中表达。检测到持家基因actg1低至5pg的uhr总rna,但未检测到乳腺癌基因,推测是因为它们在用于产生该样品的任何细胞系中不表达。
掺入血液的skbr3乳腺癌细胞
为测试测量从血液中收获的少量肿瘤细胞中mrna水平的可行性,将10或100个skbr3乳腺癌细胞掺入10ml血液中,并用基于细胞的物理特性从血细胞中部分纯化肿瘤细胞的装置,从血液中收获肿瘤细胞。然后,如上述处理来自细胞的总rna(如图1中概述)。收获的细胞主要为白细胞,在一些情况下,将仅来自白细胞的rna用作对照。
测试各个样品的六个重复:没有肿瘤细胞的血液,具有10个肿瘤细胞的10ml血液和具有100个肿瘤细胞的10ml血液(图6中各条形图从左至右布置)。除了持家基因之外,当将100个肿瘤细胞掺入10ml血液时,明确地检测到3种不同基因(erbb2、epcam和ccnd1)的mrna转录物。当以比将100个肿瘤细胞掺入血液中时低约10倍的水平,将仅10个细胞掺入10ml血液时,检测到erbb2基因的转录物。已知erbb2基因以相对高的水平在skbr3细胞中表达。
来自skbr3乳腺癌细胞的mrna定量
将图2中概述的方法用于从来自skbr3癌细胞的少量总rna中捕获mrna,并且使用导流寡核苷酸微阵列扩增和定量特异性基因。使用对基因的各个区域特异的寡核苷酸探针组,同时扩增和定量8种靶基因中每一种的3个不同区域。该24-plex测试的目的为筛选不同的寡核苷酸探针组以发现提供最大灵敏度的探针组。对于8种基因中每一种的一个探针组的结果呈现于图7中。信号强度取决于分析中使用的总rna的量。一些基因转录物仅以1皮克(pg)总rna可检测到(在log10图的轴上为零)。垂直虚线表示由单个哺乳动物细胞预期的总rna的范围(参见roozemond,histochemj.8:625,1976;uemura,brainresbull5,117,1980;brady,yeast17:211,2000)。结果表明,本公开内容的方法将能够评估单个细胞中多种mrna转录物的表达水平。
- 还没有人留言评论。精彩留言会获得点赞!
- 抑制人pard6a基因表达的小干扰核糖核酸序列及其抗卵巢癌增殖和转移的用图
- 光交联的形成抑制方法、及自交联体形成抑制型光响应性核酸的制作方法
- 糖修饰的核酸分子的制作方法
- 一种用于信息存储和加密的单链脱氧核糖核酸的制作方法
- Rnase4作为药物靶点在脑胶质瘤抑制药物中的应用
- (2S,3R,4R)-3,5-二取代-2-脱氧-2-羟基-2-甲基-D-核糖-γ-内酯的制备方法及其中间体的制作方法
- Rnase4作为药物靶点在脑胶质瘤抑制药物中的应用
- (2S,3R,4R)-3,5-二取代-2-脱氧-2-羟基-2-甲基-D-核糖-γ-内酯的制备方法及其中间体的制作方法
- 脱氧核糖核酸酶在制备用于治疗男性低生育力的药物中的应用
- 基于适体的靶向激活循环放大spr检测蛋白质的方法