一种转基因大豆检测用多重PCR试剂盒及检测方法与流程
本发明属于食品/基因检测领域,具体涉及转基因大豆检测用多重pcr试剂盒及检测方法。
背景技术:
目前,国内外报道的转基因作物检测技术主要分为三类,一类是基于核酸的检测技术;二是基于蛋白质的酶学和免疫学检测技术;第三类是转基因检测新技术,包括向自动化技术发展的生物传感器与生物芯片、基于现代分析仪器的近红外光谱和质谱分析技术等。第二类基于蛋白质的检测技术是对转基因植物中外源基因表达的蛋白进行检测,这些蛋白在加工过程中易受到破坏而失活、分解或消失,检查结果易出现假阴性且方法的重复性差。近些年发展的转基因检测新技术如生物传感器、色谱法和近红外光谱法等,此类技术目前仍处于研究阶段,存在一些暂时无法有效解决或客观存在的缺点,如色谱法只适用于转基因植物及其产品的成分发生重大变化时的定性检测分析,该类检测技术短期内仍无法有效推广应用。因此,我国农业部发布的转基因成分检测方法标准以核酸检测技术为主,该类检测技术包括pcr技术、恒温扩增技术、southern杂交、基因芯片技术、荧光定量pcr等。
多重pcr是在普通pcr的基础上,于同一个反应体系中利用多对引物同时扩增多段靶序列的pcr技术。自多重pcr报道以来,由于其高效性、灵敏性和经济简便性,已广泛应用于核酸诊断的多个领域,如遗传疾病诊断、突变和多态性分析、病原体检测、转基因鉴定等;多重pcr体系,一般包括以下试剂:pcr缓冲液、taq酶、dntp、mgcl2、多重引物、模板和去离子水。
我国目前允许进口的转基因大豆高达12种(具体包括gts-40-3-2、mon87701、mon89788、mon87701×mon89788、cv127-9、a2704-12、dp356043、dp305423、a5547、305423×40-3-2、mon87769、mon87708,其中mon87701×mon89788和305423×40-3-2为复合性状转基因大豆)。
转基因作物是利用基因工程技术将外源目的基因转入植物中培育而出的,它们具有抗虫、抗除草剂、抗逆和营养改良等优良特性。全球转基因作物的种植面积自1996年的0.17亿公顷增加到2015年的1.797亿公顷。随着转基因技术不断发展,转基因作物产业化与商业化的程度不断加深,我国陆续针对转基因作物制定了相关法律法规,然而,公众对转基因食品安全的担忧及对所食用食品是否含有转基因成分的知情权仍与日俱增,因此,快速、准确、敏感的转基因食品检测技术是所有转基因安全性评价等法律法规真正落到实处的基础和关键之一。
技术实现要素:
本发明的目的是提供一种检测转基因大豆的多重pcr试剂盒,用于我国目前批准进口(即已颁发安全证书)的12种转基因大豆的单一或者同时多重pcr检测,通过设计公共引物及转基因大豆特异性引物,限定多重pcr检测体系和条件,可检测食品(比如大豆、大豆制品、含有大豆成份的食物等)中是否含有转基因大豆成分。
为达到上述发明目的,本发明采用的技术方案是:
一种转基因大豆检测用多重pcr试剂盒,包括公共引物、针对转基因大豆的嵌合引物;所述公共引物的序列为seqidno.1;所述针对转基因大豆的嵌合引物为seqidno.2至seqidno.18所示的核苷酸序列中的一种或几种。
上述技术方案中,所述转基因大豆检测用多重pcr试剂盒还包括taq聚合酶、dntp、mgcl2、pcr缓冲液、去离子水、阳性对照引物对;其中所述阳性对照引物对的序列优选为seqidno.19、seqidno.20;可用来检测dna模板质量和反应体系扩增效率;
seqidno.19:gccctctactccacccccatcc
seqidno.20:gcccatctgcaagcctttttgtg。
上述技术方案中,进行pcr反应时,采用两步退火温度法,前10个循环退火温度为61℃;后30个循环退火温度为54℃。第一步退火温度高于第二步的退火温度5~10℃;比如前10个循环的退火温度高于后30个循环退火温度7℃,可有效提高检测的特异性;进行多重pcr反应时,针对转基因大豆的每条嵌合引物的终浓度在8~80nm之间,公共引物的终浓度为1600nm。
本发明还公开了一种检测产品中转基因大豆成分的方法,提取待检测产品基因组dna作为模板(比如转基因大豆的插入基因与大豆基因组的连接区为靶标序列),在嵌合引物与公共引物存在下采用两步退火温度法进行pcr反应,对pcr产物进行电泳分析即完成产品中转基因大豆成分的检测;最后对pcr产物扩增进行电泳分析,即完成产品中转基因大豆成分的检测;所述公共引物的序列为seqidno.1;所述嵌合引物为seqidno.2至seqidno.18所示的核苷酸序列中的一种或几种。
上述技术方案中,第一步(即前10个循环)多重pcr扩增的退火温度高于第二步(即后30个循环)多重pcr扩增的退火温度5~10℃;每条嵌合引物的终浓度在8~80nm之间,公共引物的终浓度为1600nm;所述产品为包含大豆成分的食品。具体可包括以下步骤:95℃预变性5min;95℃变性30s,61℃退火30s,72℃延伸30s,共10个循环;72℃延伸2min;95℃变性30s,54℃退火30s,72℃延伸30s,共30个循环;最后72℃延伸7min。前10个循环,在高退火温度下,低浓度的嵌合引物中特异引物扩增靶标序列,产生带有公共引物末端的pcr产物;后30个循环,在低退火温度下,高浓度的公共引物以早期产生的带有公共引物末端的pcr产物为模板进行大量扩增。根据本发明的方法可以有效、简便的检测产品中,尤其是食品中是否含有转基因大豆成分,为国民知情权以及食品安全做出巨大贡献。
本发明公开的转基因大豆成分核酸检测方法准确,可同时检测12中目标基因,而且检测结果准确,可解决公众对转基因食品安全的担忧及对所食用食品是否含有转基因成分的知情权。
本发明进一步公开了一种转基因大豆检测用引物,包括公共引物、针对转基因大豆的嵌合引物;所述公共引物的序列为seqidno.1;所述针对转基因大豆的嵌合引物为seqidno.2至seqidno.18所示的核苷酸序列中的一种或几种。
本发明还公开了上述转基因大豆检测用引物在制备检测转基因大豆的多重pcr试剂盒中的应用或者权利要求8所述转基因大豆检测用引物在检测转基因大豆中的应用。
本发明中,所述公共引物的序列seqidno.1及所述嵌合引物的核苷酸序列如下所示:
所述嵌合引物为针对12种转基因大豆的嵌合引物;a2704与a5547共用上游引物seqidno.11,mon89788、mon87769和mon87708共用下游引物seqidno.18。305423×40-3-2、mon87701×mon89788是两种基因堆叠的转基因大豆,若样品中同时检测出转基因大豆dp305423和gts40-3-2,则样品中有可能含有转基因大豆305423×40-3-2;若样品中同时检测出转基因大豆mon87701和mon89788,则样品中有可能含有转基因大豆mon87701×mon89788。
上述技术方案中,公共引物与转基因大豆相关基因组无同源性,当以转基因大豆基因组为模板进行扩增时,不论条件如何,均应没有特异性产物;当以嵌合引物的扩增产物为模板时,公共引物可以扩增出特异性产物;并且公共引物的tm比转基因大豆靶特异性引物的tm值低5~10℃。
上述技术方案中,进行pcr反应时,公共引物的终浓度为1600nm,针对转基因大豆的嵌合引物的终浓度为8~80nm,用于扩增12种转基因大豆mon87701、gts40-3-2、mon89788、cv127、a2704、a5547、dp356043、dp305423、mon87769、mon87708、305423×40-3-2、mon87701×mon89788,比如seqidno.2/3:80nm,seqidno.4/5:60nm,seqidno.6/7:64nm,seqidno.8/9:8nm,seqidno.10:32nm,seqidno.11:48nm,seqidno.12:16nm,seqidno.13/14:24nm,seqidno.15/16:8nm,seqidno.17:16nm,seqidno.18:32nm;进行pcr反应时,采用两步退火温度法,第一步退火温度比第二步退火温度高5~10℃。
本发明公开的试剂盒可用于产品尤其是食品中转基因大豆成分的检测,本发明公开的多重pcr是公共引物介导的多重pcr结合两步退火温度法。具体比如采用两步退火温度法,以转基因大豆插入基因序列与大豆基因连接区序列为靶标序列,用嵌合引物先扩增10个循环,退火温度为61℃,进行第一步多重pcr扩增,可产生公共引物末端的pcr产物;公共引物以第一步扩增的带有公共引物末端的pcr产物为模板,进行后30个循环的第二步多重pcr扩增,退火温度为54℃;即完成转基因大豆的多重pcr扩增;前10个循环的多重pcr扩增退火温度比后面30个循环的多重pcr扩增退火温度高5~10℃。循环早期,在高退火温度下,低浓度的嵌合引物扩增靶标序列,产生带有公共引物末端的pcr产物;循环后期,在低退火温度下,高浓度的公共引物以早期产生的pcr产物为模板进行大量扩增。多重pcr采用的25μl反应体系包括:10μm的公共引物4μl,不同浓度的嵌合引物混合物2μl,2×dreamtaqgreenmix12.5μl,dna模板1μl,灭菌ddh2o4.5μl;pcr反应条件为:95℃预变性5min;95℃变性30s,61℃退火30s,72℃延伸30s,共10个循环;72℃延伸2min;95℃变性30s,54℃退火30s,72℃延伸30s,共30个循环;最后72℃延伸7min。最后进行琼脂糖凝胶电泳分析,结果表明本发明的检测转基因大豆的多重pcr试剂盒可同时或者独立特异性检测10种转基因大豆的基因组;利用构建的质粒模板,本发明的检测转基因大豆的多重pcr试剂盒的敏感性最高可达0.1%(w/w)转基因大豆成分含量,即0.1ng/100ngdna样品。
由于上述技术方案运用,本发明与现有技术相比具有以下优点:
(1)本发明公开的检测转基因大豆的多重pcr试剂盒,可同时也可单独检测我国批准进口的的12种转基因大豆,即mon87701、gts40-3-2、mon89788、cv127、a2704、a5547、dp356043、dp305423、mon87769、mon87708、305423×40-3-2、mon87701×mon89788,不仅可以节约试剂使用量和降低检测成本,同时提高了检测效率,具有灵敏度高,经济、快速、操作简单等优点;
(2)本发明公开的试剂盒进行检测时,降低嵌合引物浓度,最低至8nm/每条,公共引物浓度为1600nm;降低嵌合引物的浓度有利于加入更多对嵌合引物,提高多重pcr检测通量;采用单条公共引物,有利于减少二聚体的形成,高浓度有利于扩增特异性产物的量,进而提高检测敏感性;
(3)本发明公开的试剂盒采用一个pcr反应中的两步退火温度法,前10个循环扩增采用高退火温度(61℃)有利于嵌合引物的特异性结合,减少非特异性产物;后30个循环扩增低退火温度(54℃)使公共引物发挥放大作用;中间额外增加72℃延伸2min,有利于为公共引物获得足够的模板;
(4)本发明公开的试剂盒中,公共引物与每条多重pcr特异引物的5'末端连接,一起构成了嵌合引物;公共引物介导的多重pcr可以降低引物对的浓度,减少二聚体的形成,从而提高多重pcr的检测通量;通过创造性的设计克服了现有多重pcr体系存在的非特异性扩增、易存在二聚体的缺陷;
(5)本发明的多重pcr检测体系特异性高,敏感性高达0.1%(w/w)(转基因大豆成分含量),即0.1ng/100ngdna样品;单管进行,操作简单,且具有灵敏度高、经济、快速等优点,具有实际应用价值;
(6)本发明公开的多重pcr(mutiplexpcr)在一个pcr管中同时检测多个靶标分子,比southern杂交、基因芯片方法更加简便快捷,比荧光定量pcr更经济,与恒温扩增技术(包括环介导等温扩增法)相比不易污染及高通量。由于该技术所具有的高效性、敏感性和经济简便性等优点,可以在核酸检测技术中受到广泛关注。
附图说明
图1是本发明公共引物的特异性检测电泳图;
图2是本发明公共引物敏感性结果图;
图3是本发明多重体系的特异性检测的电泳结果图;
图4是本发明检测转基因大豆测序结果图;
图5是本发明多重检测体系的敏感性结果图。
具体实施方式
下面结合实施例与附图对本发明作进一步的描述
实施例
1.公共引物的筛选及其检测的敏感性
公共引物作为整个技术体系中的核心技术,其质量好坏直接影响着技术的成败。本发明所采用的公共引物seqidno.1与转基因大豆和非转基因大豆基因组dna进行pcr扩增,在退火温度梯度45℃--65℃范围内均不能扩增特异产物,如图1,a-k为公共引物与转基因大豆和非转基因大豆基因组dna扩增结果,结果显示,公共引物均不能扩增特异产物,其中,泳道0为空白对照,泳道m为100bpdnamarker,泳道1-8为退火温度梯度45℃--65℃;a模板为非转基因大豆基因组dna,b模板为gts40-3-2基因组dna,c模板为a2704-12基因组dna,d模板为mon89788基因组dna,e模板为dp356043基因组dna,f模板为dp305423基因组dna,g模板为cv127-9基因组dna,h模板为mon87701基因组dna,i模板为a5547-127基因组dna,j模板为mon87708基因组dna,k模板为mon87769基因组dna。
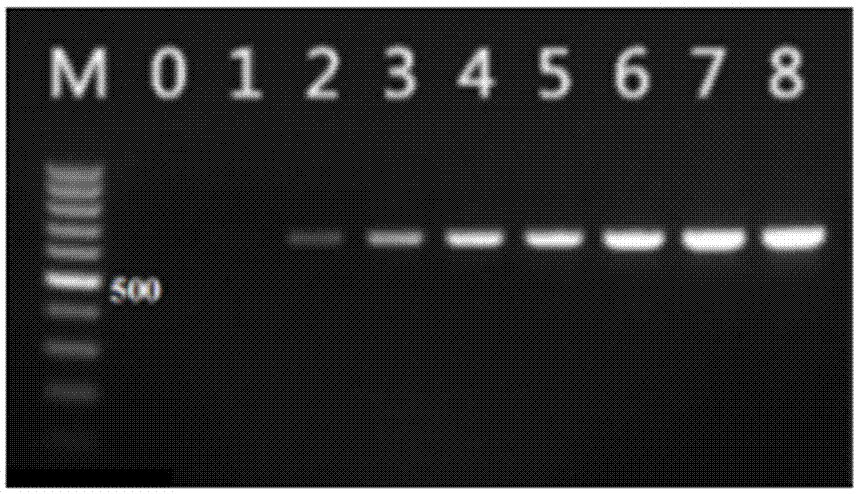
通过构建的重组质粒模板,进一步检测公共引物seqidno.1检测的敏感性,其检测敏感性达到100个拷贝,如图2,泳道m为100bpdnamarker,泳道0为空白对照,泳道1-8模板对应的质粒拷贝数依次为101、102、103、104、105、106、107、108。
本发明中,当以转基因大豆和非转基因大豆基因组dna为模板进行扩增时,不论扩增条件如何优化,均应没有扩增产物;当带有公共引物的多重嵌合引物有扩增产物时,通过公共引物一定可以扩增出特异性产物。
2.检测转基因大豆的多重pcr试剂盒,包括常规多重pcr组件,还包括针对这10种转基因大豆的嵌合引物和公共引物;嵌合引物和公共引物序列见表1,常规多重pcr组件包括taq聚合酶、dntp、mgcl2、pcr缓冲液、去离子水、阳性对照引物对;阳性对照引物对可用来检测dna模板质量和反应体系扩增效率,具体序列为:seqidno.19:gccctctactccacccccatcc、
seqidno.20:gcccatctgcaagcctttttgtg。
表1针对我国进口的12种转基因大豆嵌合引物和公共引物
3.多重体系的特异性检测
采用多引物+公共引物+单模板的pcr方法,扩增各种检测转基因大豆的基因组或靶标基因,dna模板均采用提取的相对应的10种转基因大豆基因组dna,浓度约为25ng/μl。多重pcr采用的25μl反应体系包括:10μm的公共引物4μl,不同终浓度(seqidno.2/3:80nm,seqidno.4/5:60nm,seqidno.6/7:64nm,seqidno.8/9:8nm,seqidno.10:32nm,seqidno.11:48nm,seqidno.12:16nm,seqidno.13/14:24nm,seqidno.15/16:8nm,seqidno.17:16nm,seqidno.18:32nm)的嵌合引物混合物2μl,2×dreamtaqgreenmix12.5μl,dna模板1μl,灭菌ddh2o5.5μl;pcr反应条件为:95℃预变性5min;95℃变性30s,61℃退火30s,72℃延伸30s,共10个循环;72℃延伸2min;95℃变性30s,54℃退火30s,72℃延伸30s,共30个循环;最后72℃延伸7min。图3为多重体系的特异性检测的电泳结果图,其中0为空白对照,n为阴性对照,1-10泳道是pcr产物的琼脂糖凝胶电泳结果,依次为mon87701、dp356043、dp305423、cv127、mon87769、a2704、gts40-3-2、mon87708、mon89788、a5547,n为阴性对照,m为dna标准分子量(从下到上目的带大小依次为100、200、300、400、500、600、700、800、900和1000bp),pm为产物marker,条带大小见图3上标示;结果显示,运用本发明的多重体系检测靶标dna,可以从相应模板中扩增出预期大小的特异性pcr产物,且条带清晰。扩增产物经进一步测序,证明为相对应的转基因大豆检测序列(图4)。本部分的实验结果表明,本发明的多重体系具有高特异性。
4.多重检测体系的敏感性检测
体系敏感性检测采用10种转基因大豆a2704-12、mon89788、cv127、mon87701、a5547-127、mon87708、mon87769、dp356043、dp305423、gts40-3-2基因组dna溶液等体积混合(其中dp356043、dp305423和gts40-3-2初始浓度为2.5ng/μl,其余7种初始浓度为25ng/μl),然后将此混合基因组dna溶液采用te缓冲液进行10倍梯度稀释,依次获得10、100、1000及10000倍基因组dna稀释混合液,每种稀释液取4μl作为模板(每种转基因大豆基因组dna模板浓度分别为10ng、1ng、0.1ng、0.01ng,而dp356043、dp305423、gts40-3-2这3种相对应浓度分别为1ng、0.1ng、0.01ng、0.001ng),分别对10个靶标基因进行模板梯度pcr反应,ddh2o为模板作为阴性对照。多重pcr采用25μl反应体系包括:10μm的公共引物4μl,1μm针对相应品种转基因大豆的嵌合引物对混合物2μl,2×dreamtaqgreenmix12.5μl,dna模板4μl,灭菌ddh2o2.5μl;pcr反应条件为:95℃预变性5min;95℃变性30s,61℃退火30s,72℃延伸30s,共10个循环;72℃延伸2min;95℃变性30s,54℃退火30s,72℃延伸30s,共30个循环;最后72℃延伸7min。pcr反应产物通过2%琼脂糖凝胶进行电泳,凝胶成像系统分析结果如图5,泳道m为100bpdnaladder,泳道0为去离子水为模板的空白阴性对照,1~4分别为基因组dna模板分别为稀释10000、1000、100及10倍10种转基因大豆基因组dna稀释混合液。结果显示,本发明建立的公共引物介导的多重pcr的敏感性可检测稀释1000倍10种转基因大豆基因组dna稀释混合液,体系敏感性超过0.1%,优于国家对于转基因食品检测标准要求。
sequencelisting
<110>苏州大学
<120>一种转基因大豆检测用多重pcr试剂盒及检测方法
<160>20
<210>1
<211>17
<212>dna
<213>人工合成
<400>1
ctcgtcaactccgcaag17
<210>2
<211>42
<212>dna
<213>人工合成
<400>2
ctcgtcaactccgcaagatattgaccatcatactcattgctg42
<210>3
<211>40
<212>dna
<213>人工合成
<400>3
ctcgtcaactccgcaagtcactttcttgaattagcttgct40
<210>4
<211>37
<212>dna
<213>人工合成
<400>4
ctcgtcaactccgcaagcttttgcccgaggtcgttag37
<210>5
<211>39
<212>dna
<213>人工合成
<400>5
ctcgtcaactccgcaaggccctttggtcttctgagactg39
<210>6
<211>42
<212>dna
<213>人工合成
<400>6
ctcgtcaactccgcaagcgtcaggaataaaggaagtacagta42
<210>7
<211>42
<212>dna
<213>人工合成
<400>7
ctcgtcaactccgcaagatttctaacctggctgctatagtta42
<210>8
<211>41
<212>dna
<213>人工合成
<400>8
ctcgtcaactccgcaagtgtataggaaagcgcaaactgatg41
<210>9
<211>39
<212>dna
<213>人工合成
<400>9
ctcgtcaactccgcaagattagggtttcagcaggttcgt39
<210>10
<211>43
<212>dna
<213>人工合成
<400>10
ctcgtcaactccgcaagaaaatacaaatttaacacttcattgg43
<210>11
<211>40
<212>dna
<213>人工合成
<400>11
ctcgtcaactccgcaagtaccaatgcttaatcagtgaggc40
<210>12
<211>42
<212>dna
<213>人工合成
<400>12
ctcgtcaactccgcaaggaatgcaacacactgtaacaatttg42
<210>13
<211>39
<212>dna
<213>人工合成
<400>13
ctcgtcaactccgcaagaacccttcaatttaaccgatgc39
<210>14
<211>39
<212>dna
<213>人工合成
<400>14
ctcgtcaactccgcaagttgcgaaggatagtgggattgt39
<210>15
<211>42
<212>dna
<213>人工合成
<400>15
ctcgtcaactccgcaagggcagtaacttgaaagactatgaac42
<210>16
<211>42
<212>dna
<213>人工合成
<400>16
ctcgtcaactccgcaagatgagaagatggttttttccaaggt42
<210>17
<211>43
<212>dna
<213>人工合成
<400>17
ctcgtcaactccgcaagggtaatctaaacatgcatgagaaatg43
<210>18
<211>36
<212>dna
<213>人工合成
<400>18
ctcgtcaactccgcaagtgtcgtttcccgccttcag36
<210>19
<211>22
<212>dna
<213>人工合成
<400>19
gccctctactccacccccatcc22
<210>20
<211>23
<212>dna
<213>人工合成
<400>20
gcccatctgcaagcctttttgtg23
- 还没有人留言评论。精彩留言会获得点赞!
- 一种用于检测犬副流感病毒试剂盒的制造方法与工艺
- 腹泻相关病毒多重基因检测体系及其试剂盒和应用的制造方法与工艺
- 基于POCKIT Micro荧光PCR平台的禽流感病毒H7亚型检测试剂及应用的制造方法与工艺
- 一种流行性乙型脑炎的重组酶常温扩增核酸(RT‑RPA)试纸条检测试剂盒及应用的制造方法与工艺
- 一种腺苷高半胱氨酸酶基因启动子区甲基化程度检测试剂盒及测定方法与流程
- 一种检测心肌梗死的试剂及其应用的制造方法与工艺
- 用于检测CDKN2B基因变异的试剂盒及CDKN2B基因变异率的定量检测方法与流程
- 一种CYP2C19*3基因型检测试剂盒及其检测方法与流程
- 一种CYP2C19*2基因型检测试剂盒及其检测方法与流程
- 基于高通量测序法的人类多基因突变检测试剂盒的制造方法与工艺