一种无机纳米颗粒@PAM杂化材料的制备方法与流程
本发明属于有机-无机杂化材料制备领域,具体涉及一种无机纳米颗粒@pam杂化材料的制备方法。
背景技术:
有机-无机纳米杂化材料集合了无机纳米材料和高分子材料的优点,具有柔性好、介电损耗小、易加工、介电响应等优异的性质特点,是一类具有广阔应用前景的功能材料,在转换器、探测器及非易失性存储器等电子器件中有着广泛的应用,引起了科学家的广泛关注。
随着纳米技术的快速发展,关于有机-无机纳米杂化材料的研究成果更是日新月异,很多性能优异的有机-无机纳米杂化材料被成功地开发出来。同时,也明确了制约该领域发展的两个技术瓶颈,即无机纳米粉体的分散性及其与树脂基体的相容性。
为解决上述问题,科学家们采用了很多种方法,其中最主要的方法包括:(1)采用偶联剂来处理纳米颗粒表面;(2)原位聚合法,即将纳米颗粒分散到反应单体中,再引发聚合;(3)溶胶凝胶法;(4)将拥有功能性官能团的聚合物键合到纳米颗粒表面;(5)采用表面引发聚合技术,将有机单体在纳米颗粒表面引发聚合,进而形成有机包覆膜。这些方法都有效地提高了hdpcs材料的综合性能,但都需要进一步的改进和提高。
采用纳米颗粒表面引发聚合技术,有效地继承并发展了聚合物键合技术的优点,不仅提高了有机包覆层的致密度,还能够有效地控制聚合物的厚度;可以包覆的聚合物的种类多,可以灵活地选择和设计。所以,其应用领域更加广泛,改进效果更佳,其微观结构也更可控。在该类纳米杂化材料中,无机纳米颗粒之间得到了有效的阻隔,即使在加热、混合、搅拌等条件下,仍可以有效地避免无机纳米颗粒之间的直接接触及团聚;有机物与无机物之间的相容性及键合力得到了极大的提高。研究表明该类纳米杂化材料具有高介电常数、低介电损耗、有机-无机界面结合力强的特点。
纳米颗粒表面引发聚合技术的主要工艺过程包括:(1)纳米颗粒表面处理及键合剂负载;(2)在键合剂的作用下将引发剂键合到纳米颗粒表面;(3)在一定条件下将有机单体在纳米颗粒表面引发聚合;(4)产物提纯。
在该工艺条件下,有机单体在纳米颗粒的表面被引发聚合,高分子链从纳米颗粒的表面逐渐“长出”。在聚合过程中只有单体与生长链起反应,没有明显的扩散阻碍,体积小的单体很容易到达表面活性位点和增长的聚合物链端,动力学上非常有利,因而制备的聚合物链高度伸展、择优取向、接枝密度高、分布均匀、表面覆盖度高、与纳米颗粒表面结合力强。此外,在表面引发活性聚合过程中可通过改变反应时间和条件很好地控制聚合物膜的厚度。
当前,纳米颗粒表面引发聚合机理主要包括一般自由基聚合反应、离子聚合反应及原子转移自由基聚合反应。与传统引发聚合反应(自由基聚合及离子聚合)机理相比,原子转移自由基聚合反应(atomtransferradicalpolymerization,atrp)是二十世纪90年代以来发展最为快速的自由基聚合技术,能担当合成一系列结构不同、性能特异的聚合物材料如嵌段、接枝、星状、梯状、超支化聚合物的重任。从近几年的文献报道上可以看出,纳米颗粒表面引发聚合技术中研究最多、应用最广泛、发展速度最快的就是表面引发原子转移自由基聚合。
综上所述,作为一种有效的纳米颗粒表面改性技术,表面引发聚合技术在制备高性能纳米杂化材料上有着十分广阔的应用前景,其制备工艺随着聚合理论的发展而快速进步。当前atrp等技术存在的主要问题是制备工艺复杂、反应条件苛刻,限制了其应用的深度及广度,所以开发纳米颗粒表面引发聚合的新技术和理论、简化工艺流程是该方向最需要解决的问题之一。
技术实现要素:
为解决现有技术的不足,本发明的目的在于提供一种无机纳米颗粒@pam杂化材料的制备方法。
为实现上述目的,本发明采取的技术方案如下:
一种无机纳米颗粒@pam(在无机纳米颗粒表面复合聚丙烯酰胺)杂化材料的制备方法,包括以下步骤:
s1、过氧化无机纳米颗粒的制备:
s1.1、取氢氧化钠,在冰水浴条件下边搅拌边加入到质量浓度30%的过氧化氢溶液中,得过氧化钠水溶液;其中,按质量体积比计,氢氧化钠:过氧化氢溶液=10g∶63~72ml;
s1.2、取制备的过氧化钠水溶液,加入无机纳米颗粒;其中,按质量比计,过氧化钠水溶液:无机纳米颗粒=(141.5~150)∶5;
s1.3、搅拌反应4~5h后,取出抽滤,洗涤后离心,真空干燥,得过氧化无机纳米颗粒;
s2、在s1制备的过氧化无机纳米颗粒表面聚合丙烯酰胺:
s2.1、取丙烯酰胺、水、过氧化无机纳米颗粒、氯化亚铁;其中,按质量比计,丙烯酰胺∶水∶过氧化无机纳米颗粒∶氯化亚铁=3.55∶(14~20)∶(0.02~0.03)∶(0.01~0.04);
s2.2、在氮气气氛下,60~70℃搅拌反应0.5~1h;
s2.3、反应结束后,将产物取出,加水搅拌分散,边搅拌边滴加无水乙醇至有絮状沉淀出现;
s2.4、将上层清液倒出,留下沉淀,加入无水乙醇浸泡;
s2.5、取出沉淀,真空烘干,即得无机纳米颗粒@pam杂化材料。
较好地,所述无机纳米颗粒为钛酸钡颗粒或二氧化硅颗粒。
较好地,s2.4中,加入无水乙醇浸泡两次,并且第一次加入无水乙醇浸泡0.5~1h,然后倒出无水乙醇,再加入无水乙醇开始第二次浸泡3~5h。
有益效果:
1、本发明反应体系,可以在低压加热的条件下,不需要加入或键合其他引发剂,无机纳米颗粒便可以直接将有机单体在其表面引发聚合;而且避免了制备过程中因药品本身气味等给试验人员带来的风险,也简化了制备工艺,应用的是氧化-还原体系的反应机理,使操作流程更加简便;本发明方法的主要特点:制备工艺简单,引发机理新颖;
2、本发明制备的无机纳米颗粒@pam杂化材料综合了无机纳米颗粒和pam的优点,扩宽其应用范围;钛酸钡介电性质高,pam加工温度低,钛酸钡@pam杂化材料整体既具有高的介电常数还具有很好的加工性,使得钛酸钡@pam杂化材料能够应用到电子领域,很适合嵌入式电容器(要求填入的介质材料具有高的介电常数,且基板是有机聚合物,加工温度要低)的制备。
附图说明
图1:不同过氧化时间下制备得到的过氧化钛酸钡的红外图谱。
图2:s2.4胶状沉淀(未经乙醇浸泡除去单体)的gpc图像。
图3:s2.5目标产物钛酸钡@pam杂化材料的红外图谱。
具体实施方式
以下结合实施例对本发明的技术方案作详细说明,但本发明的保护范围并不局限于此。
实施例1
过氧化钛酸钡的制备,包括以下步骤:
s1.1、取10g氢氧化钠,在冰水浴条件下边搅拌边加入到63ml质量浓度30%的过氧化氢溶液中,得过氧化钠水溶液;
s1.2、取141.5g制备的过氧化钠水溶液于200ml烧杯中,加入5g纳米级钛酸钡粉末;
s1.3、搅拌反应4h之后,取出抽滤,无水乙醇洗涤后离心,40℃真空干燥4h,得过氧化钛酸钡。
实施例2
与实施例1的不同之处在于:s1.3中,分别将搅拌反应调整为1h、2h、3h、5h、6h。
不同过氧化时间下制备得到的过氧化钛酸钡的红外图谱见图1,其中bto为原料纳米级钛酸钡粉末。在没有经过处理的钛酸钡的红外谱图上,3443cm-1为o-h的伸缩振动峰,594cm-1为ti-o的伸缩振动峰。随着反应时间的延长,在825cm-1处,出现了一个新峰,此处即为过氧化峰,随着时间的延长,峰的相对强度越大。其中在过氧化时间4h时,过氧化峰最强烈,因此选择反应时间4h制备出的过氧化钛酸钡进行下一步的实验。
实施例3
一种无机纳米颗粒@pam杂化材料的制备,即在实施例1制备的过氧化钛酸钡表面聚合丙烯酰胺,包括以下步骤:
s2.1、取3.55g丙烯酰胺、20g去离子水、0.02g过氧化钛酸钡(实施例1制备)、0.02g氯化亚铁于50ml三口烧瓶中,置于水浴锅中,同时通入氮气30min排除三口烧瓶中的空气,;
s2.2、持续通入氮气,65℃磁力搅拌反应1h;
s2.3、反应结束后,将产物取出到烧杯中,加入适量水搅拌分散,边搅拌边滴加无水乙醇至有絮状沉淀出现;
s2.4、将上层清液倒出,留下胶状沉淀,部分留样作gpc检测用,余下加入无水乙醇浸泡1h后将溶液倒出,再加入无水乙醇浸泡4h;
s2.5、取出沉淀于干燥烧杯中,40℃真空烘干4h,即得目标产物钛酸钡@pam杂化材料(bto@pam)。
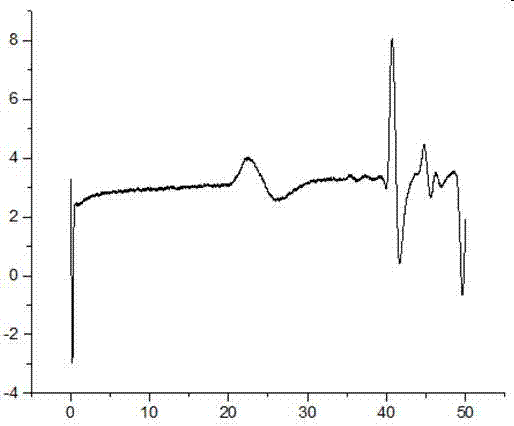
s2.4胶状沉淀(未经乙醇洗涤)的gpc图像见图2,图像横坐标为洗出时间min,纵坐标为mv黏均分子量,测试条件:色谱柱为wat044208,流动相为质量分数0.1%的叠氮化钠水溶液,流速0.8ml/min,柱温为25℃,进样量为50微升。由分子量较高的高分子先流出可知:20min时的峰表示的是高分子包覆过氧化钛酸钡,而40到50min时表示的是单体的洗出峰。
s2.5目标产物钛酸钡@pam杂化材料的红外图谱见图3。发现在图3中,3155cm-1处出现新峰,为过氧化钛酸钡和聚丙烯酰胺的复合键。
- 还没有人留言评论。精彩留言会获得点赞!