一种分泌表达蛋白的稳定细胞株构建方法与流程
本发明属于生物技术领域,特别涉及一种分泌表达蛋白的稳定细胞株构建方法。
背景技术:
构建真核表达质粒,在将其转染到293t细胞中,可以实现蛋白的瞬时表达,是目前最常用的真核蛋白表达方法。但如果需要大量表达目的蛋白就需要进行大量质粒提取、细胞培养、转染、破碎细胞提取总蛋白,再进行纯化,工作量大、纯化难度大。
将慢病毒表达载体和包装质粒同时共转染293t细胞,可以产生高滴度的病毒颗粒。再将病毒作用于宿主细胞,可以使得目的基因进入宿主细胞,经过反转录,整合到基因组,从而高水平的表达目的蛋白。再经过相应抗生素筛选即可获得稳定表达目的蛋白的细胞株,与传统质粒转染方法相比,实验周期短,阳性率高,细胞株稳定性好。
技术实现要素:
本发明的目的是解决上述现有技术的不足,提供一种分泌表达蛋白的稳定细胞株构建方法。
本发明是通过以下技术方案实现的:
一种分泌表达蛋白的稳定细胞株构建方法,包括以下步骤:
(1)以重组慢病毒载体pcdh-s-his为骨架,将目的蛋白基因构建到重组慢病毒载体pcdh-s-his上获得重组质粒pcdh-s-p-his;
(2)利用pei转染方法将上述重组质粒与辅助质粒pspax2和pmd2.g共转染293t细胞获得重组慢病毒;
(3)将收获的慢病毒转导293t细胞,经嘌呤霉素筛选获得多个单克隆细胞株,经elisa方法检测各细胞上清中目标蛋白含量,确定高表达细胞株。
优选地,步骤(2)所述重组慢病毒的获得包括以下步骤:
(21)将重组质粒pcdh-s-p-his、pspax2和pmd2.g混合后加到pbs中,轻轻混匀,同时将pei加到另一份同样体积的pbs中,涡旋混匀,再将pei混合液加到重组质粒中,轻轻混匀后室温放置10~30min;
(22)将混合液加到贴壁培养的293t细胞中;
(23)过夜或12h后换液,继续培养48h后收获上清,并补齐培养基继续培养24h收获上清,两次上清经过离心去除杂质后0.45μm滤器过滤即可获得重组慢病毒。
优选地,步骤(21)所述重组质粒pcdh-s-p-his、pspax2和pmd2.g的混合比例为质量比4:3:1,混合质粒的质量体积比浓度为16mg/l,所述pei混合液的质量体积比浓度为40mg/l。
优选地,步骤(3)所述高表达细胞株的获得包括以下步骤:
(31)将重组慢病毒加到dmem完全培养基中,同时加入基因转染增强剂polybrene;
(32)弃去293t细胞上清,加入步骤(31)的混合液,过夜培养;
(33)第二天弃去病毒液,补加dmem完全培养基,继续培养24h,荧光显微镜观察细胞绿色荧光情况(肉眼观察荧光细胞比例达到20%以上);
(34)换液48h后,弃去细胞上清,换成含不同浓度嘌呤霉素(1ug/ml-20ug/ml)的dmem完全培养基;
(35)待细胞死亡基本稳定后消化细胞进行有限稀释,接种在96孔板中,细胞数量为每孔0.5个细胞;
(36)7天后荧光显微镜观察,找到只有单细胞集落且有绿色荧光的细胞孔做好标记;
(37)待细胞密度达90%后,elisa检测阳性细胞孔上清中目标蛋白的表达量;
(38)选择高表达量的细胞株进行扩大培养,冻存。
优选地,步骤(31)所述重组慢病毒和dmem完全培养基的体积比为1:8,基因转染增强剂polybrene的终浓度为8μg/ml。
优选地,所述目的蛋白基因为人色素上皮衍生因子(pedf)基因、人胎盘生长因子(plgf)基因或成纤维生长因子受体基因(fgfr1-1α)。
优选地,所述人色素上皮衍生因子基因两端带有ecori和noti酶切位点,引物序列如seqidno.1、2所示。
优选地,所述人胎盘生长因子基因两端带有ecori和noti酶切位点,引物序列如seqidno.3、4所示。
优选地,所述成纤维生长因子受体基因两端带有bamhi和noti酶切位点,引物序列如seqidno.5、6所示。
本发明的有益效果在于:
利用本实验改造的慢病毒载体pcdh-s-his为骨架可以实现目标蛋白的分泌表达,与现有的质粒转染真核细胞获得目标蛋白相比有以下优势:包装慢病毒选择pei转染方法,试剂费用远远低于脂质体,且包装的病毒再次感染细胞效率高。经嘌呤霉素筛选出稳定表达株后可以进行大规模无血清培养,成本低,产量高,杂蛋白少,利于后期大规模纯化。本专利选取了三个基因来进行验证,均能实现目标蛋白的分泌表达,所以本专利构建分泌表达蛋白稳定株的方法具有广谱性,可以实现多数蛋白的分泌表达。但本方法表达的蛋白局限于能在293t细胞中瞬时表达的蛋白,不能解决一些在哺乳动物细胞中不表达的蛋白。本专利提供了一种分泌表达蛋白的稳定细胞株构建方法,为实现蛋白的大规模分泌表达提供可靠依据。
附图说明
图1是pcdh-cmv-mcs-ef1-copgfp-t2a-puro的图谱;
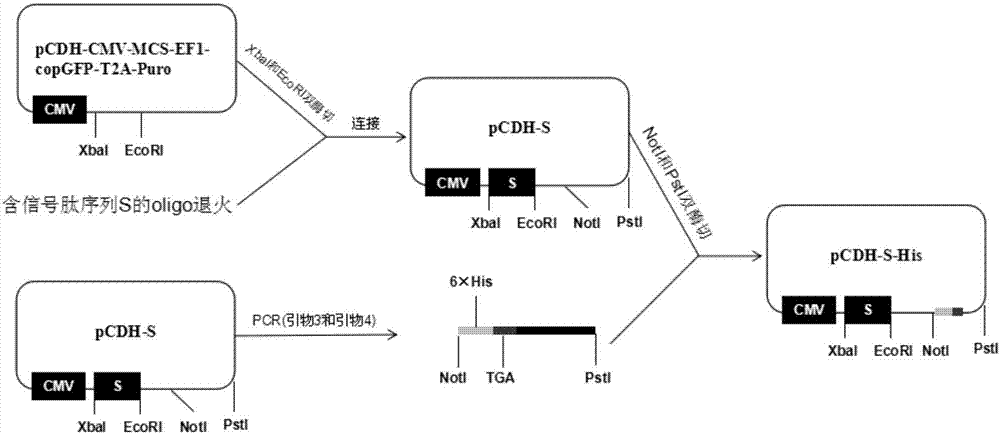
图2是pcdh-s-his重组慢病毒载体的构建流程示意图;
图3是实施例2pcdh-s-pedf-his重组质粒构建示意图;
图4是实施例3pcdh-s-plgf-his重组质粒构建示意图;
图5是实施例4pcdh-s-fgfr1-1α-his重组质粒构建示意图;
图6是实施例5重组慢病毒构建流程示意图。
具体实施方式
为更好理解本发明,下面结合实施例及附图对本发明作进一步描述,以下实施例仅是对本发明进行说明而非对其加以限定。
实施例1pcdh-s-his重组慢病毒载体的构建
将质粒pcdh-cmv-mcs-ef1-copgfp-t2a-puro(图1)进行xbai和ecori双酶切,回收载体骨架部分。如图2所示,
设计并合成2条寡核苷酸链,序列如下:
正向:(5’→3’)带有xbai粘性末端
ctagaatggagacagacacactcctgctgtgggtgctgctgctctgggtcccaggctccactggcgacggg(seqidno.7);
反向:(5’→3’)带有ecori粘性末端
aattcccgtcgccagtggagcctgggacccagagcagcagcacccacagcaggagtgtgtctgtctccata(seqidno.8)。
将上述两条寡核苷酸链退火,与载体骨架部分用连接酶进行16℃连接,1h后转化大肠杆菌过夜培养,隔天挑取单克隆菌落过夜培养,提取质粒并测序验证,最终获得重组载体pcdh-s。
重组载体pcdh-s用noti和psti双酶切,回收载体大片段部分。
根据重组载体pcdh-s序列设计引物,以重组载体pcdh-s的noti酶切位点处设计上游长引物,并包含6×his标签,并添加终止密码子,以psti酶切位点处设计下游引物,引物序列如下:
上游引物:
5’-tccgcggccgctcatcatcaccatcaccattgagaaggatctgcgatcgct-3’(seqidno.9);
下游引物:
5’-ttctgcaggatgctggggtggat-3’(seqidno.10)。
以重组载体pcdh-s为模板,通过pcr反应获得pcr产物。扩增程序为预变性95℃4分钟,变性95℃30秒,退火55℃30秒,延伸72℃1分钟,循环数35个,最后延伸72℃10分钟。
将pcr产物纯化后用,经noti和psti双酶切,跑胶回收,再与酶切回收的pcdh-s载体大片段用连接酶进行连接转化,挑取单克隆菌落培养后提取质粒并测序验证,最终获得改造后的重组慢病毒载体pcdh-s-his。
实施例2pcdh-s-pedf-his重组质粒的构建
如图3所示,
以重组慢病毒载体pcdh-s-his为骨架,经ecori和noti双酶切回收骨架部分。
设计引物扩增pedf基因,引物序列如下:
上游引物:
5’-tttgaattctatgcaggccctggtgcta-3’(seqidno.1);
下游引物:
5’-tttgcggccgcggggcccctggggtccagaa-3’(seqidno.2);
以人cdna为模板,通过pcr反应获得pedf基因。扩增程序为预变性95℃4分钟,变性95℃30秒,退火55℃30秒,延伸72℃1分钟20秒,循环数35个,最后延伸72℃10分钟。
将pcr产物纯化后用ecori和noti双酶切,跑胶回收,再与酶切回收的pcdh-s-his载体骨架部分用连接酶进行连接,再转化大肠杆菌,挑取单克隆菌落培养后提取质粒并测序验证。最终获得重组质粒pcdh-s-pedf-his。
实施例3pcdh-s-plgf-his重组质粒的构建
如图4所示,
以重组慢病毒载体pcdh-s-his为骨架,经ecori和noti双酶切回收骨架部分。
设计引物扩增plgf基因,引物序列如下:
上游引物:
5’-tttgaattccctgcctgctgtgccccccca-3’(seqidno.3);
下游引物:
5’-tttgcggccgcgcctccggggaacagcat-3’(seqidno.4);
以人cdna为模板,通过pcr反应获得plgf基因。扩增程序为预变性95℃4分钟,变性95℃30秒,退火55℃30秒,延伸72℃1分钟20秒,循环数35个,最后延伸72℃10分钟。
将pcr产物纯化后用ecori和noti双酶切,跑胶回收,再与酶切回收的pcdh-s-his载体骨架部分用连接酶进行连接,再转化大肠杆菌,挑取单克隆菌落培养后提取质粒并测序验证。最终获得重组质粒pcdh-s-plgf-his。
实施例4pcdh-s-fgfr1-1α-his重组质粒的构建
如图5所示,
以重组慢病毒载体pcdh-s-his为骨架,经bamhi和noti双酶切回收骨架部分。
设计引物扩增fgfr1-1α基因,引物序列如下:
上游引物:
5’-cgggatcctgaggccgtccccgaccttgcctgaac-3’(seqidno.5);
下游引物:
5’-ataagaatgcggccgccgaggtcatcactgccggcctc-3’(seqidno.6);
以人cdna为模板,通过pcr反应获得fgfr1-1α基因。扩增程序为预变性95℃4分钟,变性95℃30秒,退火55℃30秒,延伸72℃1分钟20秒,循环数35个,最后延伸72℃10分钟;
将pcr产物纯化后分别用bamhi和noti双酶切,跑胶回收,再与酶切回收的pcdh-s-his载体骨架部分用连接酶进行连接,再转化大肠杆菌,挑取单克隆菌落培养后提取质粒并测序验证。最终获得重组质粒pcdh-s-fgfr1-1α-his。
实施例5病毒包装
以pedf为例,如图6所示,
质粒大提:将重组质粒pcdh-s-pedf-his,两个辅助质粒pspax2和pmd2.g进行大提获得高浓度高纯度无内毒素的质粒。
细胞培养:以常规方法培养293t细胞,稳定传代后接种细胞至60mm培养皿,细胞数量以培养24小时达85%汇合度为依据;
将质粒pcdh-s-pedf-his、pspax2和pmd2.g按照4:3:1比例共8μg质粒加到500μlpbs中,轻轻混匀,同时将20μgpei加到500μlpbs中,涡旋混匀。再将pei混合液加到质粒中,轻轻混匀后室温放置20min;
先吸走293t细胞的培养基1ml,再将混合液加到细胞中;
过夜培养或待12h后换液,继续培养48h后收获上清,同时再补加新鲜培养基继续培养24h后收获上清。期间在荧光显微镜下观察细胞绿色荧光情况,确保转染过程正常,转染效率正常;
将两次上清混合后4℃10000g离心10min,收集上清;
将上清用0.45μm滤器过滤,分装病毒液并低温冻存。
实施例6病毒转导293t细胞筛选稳定细胞株
细胞接种:以常规方法培养293t细胞,并接种一个60mm培养皿;
将500μl重组慢病毒加到4mldmem完全培养基中,同时加入polybrene(终浓度为8μg/ml);
弃去60mm培养皿细胞上清,加上上述混合液,过夜培养;
换液后继续培养24h,荧光显微镜观察细胞绿色荧光比例;
再次换液,培养基中添加嘌呤霉素;
待细胞死亡基本稳定,胰酶消化细胞进行有限稀释,接种4块96孔板,细胞数量为每孔0.5个细胞;
7天后荧光显微镜观察,找到只有单细胞集落且有绿色荧光的细胞孔做好标记;
待细胞长满后收集细胞上清,elisa检测阳性细胞孔上清中目标蛋白的表达量,elisa结果见表1。其中pedf蛋白的b6和b11号细胞,plgf蛋白的f6和f11号细胞,fgfr1-1α蛋白的e10和h11号细胞为高表达量稳定细胞株,分别进行扩大培养,冻存。
表1稳定株上清目标蛋白elisa检测
以上所述实施方式仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案作出的各种变形和改进,均应落入本发明的权利要求书确定的保护范围内。
sequencelisting
<110>合肥知恩生物技术有限公司
<120>一种分泌表达蛋白的稳定细胞株构建方法
<130>2
<160>10
<170>patentinversion3.3
<210>1
<211>28
<212>dna
<213>人工合成
<400>1
tttgaattctatgcaggccctggtgcta28
<210>2
<211>31
<212>dna
<213>人工合成
<400>2
tttgcggccgcggggcccctggggtccagaa31
<210>3
<211>30
<212>dna
<213>人工合成
<400>3
tttgaattccctgcctgctgtgccccccca30
<210>4
<211>29
<212>dna
<213>人工合成
<400>4
tttgcggccgcgcctccggggaacagcat29
<210>5
<211>35
<212>dna
<213>人工合成
<400>5
cgggatcctgaggccgtccccgaccttgcctgaac35
<210>6
<211>38
<212>dna
<213>人工合成
<400>6
ataagaatgcggccgccgaggtcatcactgccggcctc38
<210>7
<211>71
<212>dna
<213>人工合成
<400>7
ctagaatggagacagacacactcctgctgtgggtgctgctgctctgggtcccaggctcca60
ctggcgacggg71
<210>8
<211>71
<212>dna
<213>人工合成
<400>8
aattcccgtcgccagtggagcctgggacccagagcagcagcacccacagcaggagtgtgt60
ctgtctccata71
<210>9
<211>51
<212>dna
<213>人工合成
<400>9
tccgcggccgctcatcatcaccatcaccattgagaaggatctgcgatcgct51
<210>10
<211>23
<212>dna
<213>人工合成
<400>10
ttctgcaggatgctggggtggat23
- 还没有人留言评论。精彩留言会获得点赞!