一种治疗癌症的药物水合物及其制备方法与流程
本发明属于医药
技术领域:
,公开了一种治疗癌症的药物水合物及其制备方法,具体涉及一种奥拉帕尼三水合物及其制备方法。
背景技术:
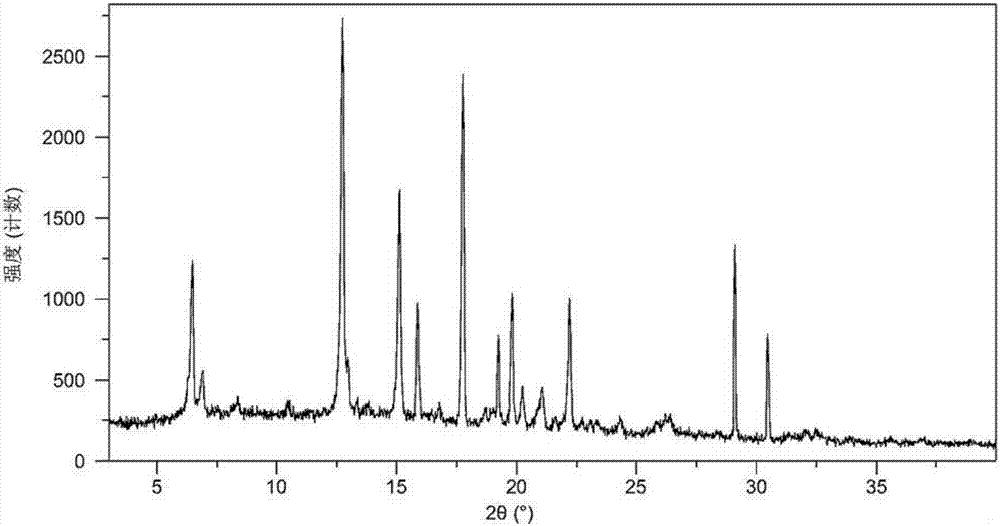
:奥拉帕尼,化学名为4-[3-(4-环丙烷羰基哌嗪-1-羰基)-4-氟苄基]-2h-酞嗪-1-酮,英文名为olaparib,结构式如式1所示:奥拉帕尼(olaparib)是由阿斯利康(astrazeneca)的全资子公司kudos制药公司研发的一种小分子,是一种强效parp抑制剂,它通过抑制肿瘤细胞dna损伤修复,促进肿瘤细胞凋亡,从而可增强放疗以及烷化剂和铂类药物化疗的疗效,主要用于治疗乳腺癌基因一号或二号(brca-1或brca-2)的基因突变癌(主要存在于乳腺癌,卵巢癌和前列腺癌)。奥拉帕尼可以选择性的作用于肿瘤细胞,正常细胞由于保留了双链修复功能而不被破坏。癌细胞由于两个等位基因都缺失或突变,双链修复功能丧失,细胞最终死亡,这一新型的药物给难治性肿瘤的治疗带来了希望。cn101528714a公开了奥拉帕尼无溶剂化物的晶型a,其x-射线粉末衍射图(2θ)在12.0°、17.8°、21.1°、22.3°、29.2°、10.5°、14.0°、21.7°、24.3°、26.1°存在特征峰,dsc显示在210.1℃±1℃存在吸热峰,该晶型稳定性好,经检测其为无水合物。专利cn105439961a公开了奥拉帕尼的晶型i及其制备方法,本发明提供的晶型i,其特征在于,其x射线粉末衍射图在2theta值为6.4°±0.2°、12.7°±0.2°、15.1°±0.2°处具有特征峰,经检测其含有1.5分子的结晶水。其制备方法为:将奥拉帕尼的固体置于纯水或含水溶剂中搅拌得到,所述含水溶剂,包括含水体积不小于80%的混合溶剂,所述含水溶剂,包括醇类、酮类、醚类、烷烃类、芳香类溶剂。该晶型稳定性比专利cn101528714a中的晶型a更好,在制备、储存以及制剂开发过程中都能够保持稳定,不会发生转晶;溶解度、引湿性符合药用要求,且制备方法所用溶剂无毒环保,对未来该药物的优化和开发具有重要价值,为药物固体制剂提供一个更好的选择。专利cn105254572a公开了一种奥拉帕尼的晶型及其制备方法,使用cukα辐射,以2θ角度表示的粉末x射线衍射在22.9±0.5,23.4±0.5,21.0±0.5,17.1±0.5,17.3±0.5,14.2±0.5,15.0±0.5,13.5±0.5,18.6±0.5,20.6±0.5,10.2±0.5,20.3±0.5,21.9±0.5,25.8±0.5,26.5±0.5有衍射峰,经检测其为无水合物。其表现为当升温速率为每分钟10℃的dsc图谱中存在1个吸热峰在175.3℃±1℃处。本发明还提供了奥拉帕尼晶型的制备方法,包括:1)将奥拉帕尼与溶剂混合回流,得到奥拉帕尼溶液,所述溶剂为正丙醇、异丙醇、正丁醇、异丁醇和叔丁醇中的一种或几种;2)将奥拉帕尼溶液过滤,自然冷却,得到所述的奥拉帕尼晶型。本发明提供的晶型奥拉帕尼的粒度小,溶出速度快。本发明所述的晶型奥拉帕尼的粒度为d50为2.45微米,粒度较小,进而使得其作为制剂时,溶出速度快,提高了产品的药效;且本发明提供的制备方法简单,得到的晶型奥拉帕尼性能也稳定。cn101821242a公开了奥拉帕尼无溶剂化物的晶型l,其x-射线粉末衍射图(2θ)在14.4°、17.2°、17.5°、18.8°、23.0°、10.4°、13.6°、25.1°存在特征峰,dsc显示在198.5℃±1℃存在吸热峰,该晶型稳定性好,经检测其为无水合物。cn106554315a公开了一种奥拉帕尼一水合物,其x-射线粉末衍射图(2θ)在5.620°、6.420°、7.420°、9.340°、10.120°、11.240°、14.020°、16.880°、17.540°、18.120°、20.200°、22.460°、24.180°、26.720°、30.200°、33.100°、34.640°、37.340°、39.420°存在特征峰,其制备方法为奥拉帕尼加入4-5倍重量/体积比丙酮-水=3-4:2-1的混合液中,加热至70-75℃,趁热过滤,滤液自然冷却至室温,再保温5-10小时,析出结晶,过滤,经干燥得到。本发明的晶体具有的优点:纯度高,稳定性好,即使在高湿度条件下吸湿增重也不明显。奥拉帕尼属于难溶性药物,常常制备成固体制剂给药,而对于晶型药物的固体制剂来说,制剂的稳定性以及溶出度与原料药的晶型有很大的关系,奥拉帕尼在结晶时,如果采用不同的溶剂和工艺条件,则其分子在各晶型晶胞的排列数目和位置及点阵形式不一样,形成不同的晶体结构,奥拉帕尼多晶型的变化会改变其性质,质量和药效。因此,奥拉帕尼的稳定结晶,对于进一步研究该化合物的物理化学性质,研究其药物组合及临床应用,具有十分重要的意义。化合物多晶型是一种普遍现象,对于晶型的稳定性、吸湿性、溶解性等优良性能一直是一种长远的追求。发明经过大量的试验研究发现,现有晶型大多数从稳定性、吸湿性解决其制剂的稳定问题,而制剂的溶出则常常是通过制剂过程中辅料的添加及原料晶型的控制等角度解决。奥拉帕尼原料的难溶性一直是本领域的技术难点,一直无法突破。本发明经过大量的试验研究,采用新的结晶方法,得到了一种新的奥拉帕尼晶型,经检测其为三水合物,本发明提供的奥拉帕尼三水合物纯度高、稳定性好,经实验惊喜地发现本发明得到的奥拉帕尼三水合物溶解性显著提高。本发明还公开了奥拉帕尼三水合物的制备方法,该制备方法简单易操作,反应条件温和,收率及纯度高,适合大规模生产。本发明的奥拉帕尼三水合物制成的胶囊溶出度及稳定性显著提高,非常适合临床应用。技术实现要素:本发明旨在提供一种奥拉帕尼三水合物及其制备方法,所要解决的技术问题是提高奥拉帕尼在水中的溶解性和纯度,从而提高其制剂的溶出度和生物利用度,以克服现有技术缺陷。为了实现本发明的目的,采用的技术方案为:一种治疗癌症的药物水合物,该水合物为奥拉帕尼三水合物,其分子式为:c24h23fn4o3·3h2o,结构式如下(式ii),其以2θ±0.2°衍射角表示的x-射线粉末衍射图谱在6.3928°、6.9115°、12.6931°、15.1126°、15.9045°、17.7123°、19.2086°、19.7743°、20.2325°、21.0346°、22.2218°、29.0633°、30.4218°处显示有特征衍射峰。本发明提供的奥拉帕尼三水合物使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示。本发明还提供了一种奥拉帕尼三水合物的制备方法,具体步骤为:(1)取奥拉帕尼粗品,加入四甲基乙二胺/水的混合溶液,加热搅拌溶解1-2min,活性炭脱色,抽滤;(2)将步骤(1)滤液降至-2℃,滴加预冷的四甲基乙二胺,继续降温析晶;(3)保温搅拌至析晶完全,养晶,抽滤,水洗,干燥,得白色结晶性粉末。优选地,步骤(1)所述的四甲基乙二胺/水混合溶液中,四甲基乙二胺与水的体积比为25:1.0~1.2;步骤(1)所述奥拉帕尼粗品与混合溶液的质量体积比为1g:10ml~12ml;所述的搅拌速度为30-50转/分钟。进一步优选地,步骤(1)所述的四甲基乙二胺/水混合溶液中,四甲基乙二胺与水的体积比为25:1.1;步骤(1)所述奥拉帕尼粗品与混合溶液的质量体积比为1g:11ml;所述的搅拌速度为40转/分钟。优选地,步骤(2)所述四甲基乙二胺与步骤(1)混合溶液的体积比0.5~1:1;步骤(2)所述滴加速度为1.0~2.0ml/min,降温幅度为每10分钟1℃~3℃;步骤(2)所述降温析晶为降温至-10℃~-8℃析晶。进一步优选地,步骤(2)所述四甲基乙二胺与步骤(1)混合溶液的体积比0.8:1;步骤(2)所述滴加速度为1.5ml/min,降温幅度为每10分钟2℃;步骤(2)所述降温析晶为降温至-9℃析晶。优选地,步骤(3)所述养晶时间为1h~3h;步骤(3)所述干燥温度为40℃~50℃。进一步优选地,步骤(3)所述养晶时间为2h;步骤(3)所述干燥温度为45℃。本发明中,所述的奥拉帕尼粗品可以为待进一步纯化的奥拉帕尼固体混合物奥拉帕尼粗品也可以为市售原料或通过现有技术方法制备得到,所得的晶型结果均在误差范围内,均为本发明新晶型。本发明还提供了一种含有本发明奥拉帕尼三水合物的药物组合物,该药物组合物为含有奥拉帕尼三水合物的胶囊。晶体的形成机理很复杂,一个新晶体的获得也具有很大的偶然性,有时不同的溶剂、在不同的结晶条件下会产生相同的晶体结构。某些特定晶型也并非一定会获得更加有利的理化性质。药物的稳定性、吸湿性、溶解性、生物活性、毒性等性质会因晶型的不同而产生巨大的差异。本发明经过大量的试验筛选,通过选择不同的溶剂溶解及不同的溶剂析晶,得到了本发明的制备方法,通过对搅拌速度、溶剂用量、温度及养晶时间的控制,意外的得到了一种奥拉帕尼新晶型,经检测其为三水合物。本发明提供的奥拉帕三水合物尼纯度高、稳定性好,经实验惊喜地发现本发明得到的奥拉帕尼三水合物溶解性显著提高。本发明还公开了奥拉帕尼三水合物的制备方法,该制备方法简单易操作,反应条件温和,适合大规模生产。本发明的奥拉帕尼三水合物制成的胶囊溶出度及稳定性显著提高,非常适合临床应用。研究表明,在x-射线粉末衍射图谱中,由新晶型得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。本发明所提供的奥拉帕尼三水合物结晶证实含有三分子的结晶水,其性状为白色结晶性粉末,在常温干燥条件下不会发生结晶水的流失。且其粉末x-射线衍射图谱与现有技术具有明显不同的峰的相对位置,可见其是一种与现有技术不同的新晶型。下面通过对本发明提供的奥拉帕尼三水合物晶型进行研究来解释和说明本发明技术方案:1、元素分析c24h23fn4o3·3h2o仪器:varioelcube元素分析仪;测量元素为c、h、o、n;dioenxdx-500型离子色谱仪;测量元素为f。元素分析(%)理论值为:h(5.98),c(59.01),n(11.47),o(19.65),f(3.89)。元素分析(%)测定值为:h(5.99),c(59.02),n(11.45),o(19.64),f(3.90)。与元素分析的理论值基本相符。2、晶型检测取本发明制备得到的奥拉帕尼三水合物,使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示,其以2θ±0.2衍射角表示的x-射线粉末衍射图在6.3928°、6.9115°、12.6931°、15.1126°、15.9045°、17.7123°、19.2086°、19.7743°、20.2325°、21.0346°、22.2218°、29.0633°、30.4218°处显示有特征峰。3、差热分析及热重分析对本发明制备的奥拉帕尼三水合物进行热重和差热分析,结果如附图2和附图3所示;结果表明,本品在100℃左右快速失去约3个水分子的重量;本品在约210℃处有吸热峰,从侧面证明了其为一种不同的晶型。4、水分分析采用卡式水分测定仪测定,本发明的奥拉帕尼三水合物的水含量为11.06-11.08%,与三水合物的理论含水量11.06%一致,证明本发明含3分子的结晶水。5、纯度检测经hplc纯度检测,本发明制备得到的奥拉帕尼三水合物的纯度可达到99.97~99.99%。6、熔点检测取本发明制备得到的奥拉帕尼三水合物进行检测,熔点为209~211℃。与现有技术相比,本发明具有如下优点:(1)本发明所提供的奥拉帕尼三水合物是一种不同于现有技术的新晶型;本发明所提供的奥拉帕尼三水合物的制备方法简单易操作,反应条件温和,收率大于99.8%,纯度高,适合大规模生产。(2)本发明提供的奥拉帕尼三水合物纯度高、稳定性好,经实验惊喜地发现本发明得到的奥拉帕尼三水合物溶解性显著提高。本发明的奥拉帕尼三水合物制成的胶囊溶出度及稳定性显著提高,非常适合临床应用。附图说明图1为本发明制备的奥拉帕尼三水合物的x-射线粉末衍射图谱。图2为本发明实施例1制备的奥拉帕尼三水合物的热重分析tga图谱。图3为本发明实施例1制备的奥拉帕尼三水合物的dsc图谱。具体实施例以下用实施例对本发明的技术方案进行详细说明,将有助于对本发明的技术方案的优点、效果有更进一步的了解,实施例不限定本发明的保护范围,本发明的保护范围由权利要求来决定。实施例1:奥拉帕尼三水合物的制备(1)取奥拉帕尼粗品100g,加入四甲基乙二胺/水(体积比为25:1.0)的混合溶液1000ml,加热搅拌(30转/分钟)溶解1-2min,活性炭脱色,抽滤;(2)将步骤(1)滤液以每10分钟1℃的速度降至-2℃,以1.0ml/min的速度滴加预冷的四甲基乙二胺500ml,继续以每10分钟1℃的速度降温至-10℃析晶;(3)保温搅拌至析晶完全,养晶1h,抽滤,水洗,40℃干燥,得白色结晶性粉末99.90g,收率99.90%,纯度99.98%。所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图见图1。实施例2:奥拉帕尼三水合物的制备(1)取奥拉帕尼粗品100g,加入四甲基乙二胺/水(体积比为25:1.2)的混合溶液1200ml,加热搅拌(50转/分钟)溶解1-2min,活性炭脱色,抽滤;(2)将步骤(1)滤液以每10分钟3℃的速度降至-2℃,以2.0ml/min的速度滴加预冷的四甲基乙二胺1200ml,继续以每10分钟3℃的速度降温至-10℃析晶;(3)保温搅拌至析晶完全,养晶3h,抽滤,水洗,50℃干燥,得白色结晶性粉末99.89g,收率99.89%,纯度99.97%。所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。实施例3:奥拉帕尼三水合物的制备(1)取奥拉帕尼粗品100g,加入四甲基乙二胺/水(体积比为25:1.1)的混合溶液1100ml,加热搅拌(40转/分钟)溶解1-2min,活性炭脱色,抽滤;(2)将步骤(1)滤液以每10分钟2℃的速度降至-2℃,以1.5ml/min的速度滴加预冷的四甲基乙二胺880ml,继续以每10分钟2℃的速度降温至-9℃析晶;(3)保温搅拌至析晶完全,养晶2h,抽滤,水洗,45℃干燥,得白色结晶性粉末99.95g,收率99.95%,纯度99.99%。所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。实施例4:奥拉帕尼三水合物的制备(1)取奥拉帕尼粗品100g,加入四甲基乙二胺/水(体积比为25:1.1)的混合溶液1200ml,加热搅拌(40转/分钟)溶解1-2min,活性炭脱色,抽滤;(2)将步骤(1)滤液以每10分钟1℃的速度降至-2℃,以1.0ml/min的速度滴加预冷的四甲基乙二胺960ml,继续以每10分钟2℃的速度降温至-8℃析晶;(3)保温搅拌至析晶完全,养晶2h,抽滤,水洗,45℃干燥,得白色结晶性粉末99.92g,收率99.92%,纯度99.98%。所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。实施例5:奥拉帕尼三水合物的制备(1)取奥拉帕尼粗品100g,加入四甲基乙二胺/水(体积比为25:1.0)的混合溶液1000ml,加热搅拌(50转/分钟)溶解1-2min,活性炭脱色,抽滤;(2)将步骤(1)滤液以每10分钟1℃的速度降至-2℃,以1.5ml/min的速度滴加预冷的四甲基乙二胺1000ml,继续以每10分钟3℃的速度降温至-10℃析晶;(3)保温搅拌至析晶完全,养晶3h,抽滤,水洗,50℃干燥,得白色结晶性粉末99.93g,收率99.93%,纯度99.99%。所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。实施例6:奥拉帕尼三水合物的制备(1)取奥拉帕尼粗品100g,加入四甲基乙二胺/水(体积比为25:1.2)的混合溶液1200ml,加热搅拌(30转/分钟)溶解1-2min,活性炭脱色,抽滤;(2)将步骤(1)滤液以每10分钟3℃的速度降至-2℃,以2.0ml/min的速度滴加预冷的四甲基乙二胺1200ml,继续以每10分钟2℃的速度降温至-9℃析晶;(3)保温搅拌至析晶完全,养晶1h,抽滤,水洗,45℃干燥,得白色结晶性粉末99.92g,收率99.92%,纯度99.98%。所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。下面通过实验例进一步说明本发明:实验例1:溶剂筛选试验采用本发明的制备方法操作,具体如下:(1)取奥拉帕尼粗品100g,加入1000ml溶剂a/溶剂b的混合溶液,加热搅拌(30转/分钟)溶解1-2min,活性炭脱色,抽滤;(2)将步骤(1)滤液以每10分钟3℃的速度降至-2℃,以1.0ml/min的速度滴加预冷的1000ml溶剂c,继续以每10分钟1℃的速度降温至-10℃析晶;(3)保温搅拌至析晶完全,养晶2h,抽滤,水洗,40℃干燥,得白色结晶性粉末。表1溶剂筛选实验结果发明人在溶剂筛选过程中,对大多数有机溶剂进行了筛选,不同的组合结晶效果不同,在此仅列举部分筛选试验的数据。发明人在试验过程中惊喜地发现在四甲基乙二胺/水混合溶剂体系溶解奥拉帕尼粗品,并用四甲基乙二胺析晶,效果相对于单用其他溶剂体系更好。然后进一步对四甲基乙二胺/水的混合溶剂的比例进行了筛选,发现当四甲基乙二胺/水的体积比为25:1.0~1.2时,效果是最好的,不仅纯度高、收率高,而且通过实验惊喜地发现其溶解性显著提高。故最终确定选择以四甲基乙二胺/水25:1.0~1.2为溶解溶剂,加四甲基乙二胺析晶,以做进一步筛选。实验例2:结晶试验条件筛选(1)取奥拉帕尼粗品100g,加入四甲基乙二胺/水的混合溶液,加热搅拌溶解1-2min,活性炭脱色,抽滤;(2)将步骤(1)滤液降至-2℃,滴加预冷的四甲基乙二胺,继续降温析晶;(3)保温搅拌至析晶完全,养晶,抽滤,水洗,干燥,得白色结晶性粉末。表2-1结晶试验条件筛选结果表2-2结晶试验条件筛选结果表2-3结晶试验条件筛选结果表2-4结晶试验条件筛选结果关于结晶试验条件的筛选试验非常复杂,在此我们只列举其中的一部分试验结果。从上述试验结果可以看出,晶体的结晶过程中存在太多的变量,每一个工艺参数的变化都可能对结果产生影响。发明人经过大量试验,最终确定了本发明技术方案的工艺。实验例3:溶解度测定试验品:本发明实施例1-6所制备的样品;对照品1:参照专利cn105439961a实施例1、2制备的奥拉帕尼晶型i。对照品2:参照专利cn105254572a实施例1-3制备的奥拉帕尼晶型。对照品3:参照专利101528714a实施例1、3、5制备的奥拉帕尼晶型a。对照品4:参照专利cn101821242a实施例1-5制备的奥拉帕尼晶型l。对照品5:参照专利cn106554315a实施例1制备的奥拉帕尼一水合物晶型化合物。对照品6:参照专利cn105985294a实施例1-4制备的奥拉帕尼。对照品7:参照专利cn105061328a实施例1-4制备的奥拉帕尼。对照品8:参照专利cn105085407a实施例一、二、三制备的奥拉帕尼。对照品9:参照专利cn105503739a实施例1-4制备的奥拉帕尼。对照品10:参照专利cn105820126a实施例4-1、4-2、4-3制备的奥拉帕尼。对照品11:参照文献j.am.chem.soc.2014,136,6142-6147制得的奥拉帕尼。对照品12:参照文献j.med.chem.,2008,51:6581-6591制得的奥拉帕尼。对照品13:参照南京工业大学2012年硕士论文《奥拉帕尼及其类似物合成研究》报道的方法制得的奥拉帕尼。上述对照品均为经过多次重复实验制得的晶型,并多次进行x射线粉末衍射检测,待晶型稳定(检测结果基本一致)方可作为对照品(如原专利公开了附图,则将测定图与之对比,基本一致方可作为对照品使用)。参照中国药典2015年版二部凡例测定其溶解性,方法:取本品适量,分别加入水,每隔5分钟强力振摇30秒钟,观察30分钟内的溶解情况,即得,结果见表3。表3本发明的晶型和对照品在水中溶解性试验结果将上述实施例1-6溶解的水溶液样品在25℃恒温搅拌72小时,取样5ml。样品经0.45μm微孔滤膜过滤,弃去初滤液,取续滤液20μl测定药物含量即为水中溶解度(mg/ml)。结果见表4:表4本发明晶型与现有技术晶型在水中的溶解度对比从上表可以看出,25℃下,本发明奥拉帕尼新晶型的在水中的溶解度与现有技术相比,有显著提高,取得了意料不到的技术效果。实验例4:稳定性试验实验例通过加速试验和长期试验,考察本发明提供的奥拉帕尼结晶的稳定性。1、加速试验取实施例1-3制备的样品,于温度40±2℃、相对湿度75±5%的条件下放置6个月,分别于0、1、2、3、6个月末取样测定性状、有关物质、含量,结果见表5。表5:加速试验结果(温度40±2℃,相对湿度75±5%)从表5看出,本发明奥拉帕尼结晶在温度40±2℃、相对湿度75±5%的条件下放置6个月,有关物质含量没有明显升高,各指标均无明显变化,含水量稳定(3分子水),从另一方面证明了该化合物中的水是结晶水不是吸附水。2、长期试验取实施例1-3制备的样品,于温度25±2℃、相对湿度60±5%的条件下放置6个月,分别于0、3、6、9、12、18、24个月末取样测定性状、有关物质、含量,结果见表6。表6:长期试验结果(温度25±2℃,相对湿度60±5%)从表6看出,本发明奥拉帕尼结晶在温度25±2℃、相对湿度60±5%的条件下放置24个月稳定,各指标均无明显变化,含水量稳定(3分子水),从另一方面证明了该化合物中的水是结晶水不是吸附水。其他实施例加速和长期试验检测结果一致。实验例5:胶囊剂溶出度检测参照专利cn106551916a实施例1的处方及工艺制备胶囊剂,所不同的是选择不同的晶型化合物,分别制备得到胶囊剂,对得到的胶囊剂进行溶出度检测,结果见表7。溶出检测方法:采用篮法,在37℃和100rpm搅拌速度下,置于900ml的0.3%sds溶液中。在90分钟后,取1ml样品,并通过hplc检测奥拉帕尼含量。表7溶出度检测结果样品溶出(%)样品溶出(%)实施例1晶型制得的胶囊99.2对照品5晶型制得的胶囊97.8实施例2晶型制得的胶囊99.3对照品6晶型制得的胶囊98.0实施例3晶型制得的胶囊99.3对照品7晶型制得的胶囊98.1实施例4晶型制得的胶囊99.4对照品8晶型制得的胶囊98.0实施例5晶型制得的胶囊99.3对照品9晶型制得的胶囊97.7实施例6晶型制得的胶囊99.1对照品10晶型制得的胶囊97.9对照品1晶型制得的胶囊98.1对照品11晶型制得的胶囊98.6对照品2晶型制得的胶囊97.5对照品12晶型制得的胶囊98.0对照品3晶型制得的胶囊97.2对照品13晶型制得的胶囊98.1对照品4晶型制得的胶囊98.2由上述检测结果可以看出,采用本发明晶型制得的胶囊剂比现有晶型制得的胶囊剂溶出度显著提高。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1