蛋白酶响应性线形-树枝状嵌段共聚物及其制法和用途的制作方法
本发明属于功能高分子材料领域,具体涉及对蛋白酶有响应特性的、以聚(羟乙基-l-谷氨酰胺)(pheg)为线形链的线形-树枝状嵌段共聚物、其制备方法和用途。
背景技术:
由线形高分子与树枝状大分子(树枝化基元)通过化学键连接而成的线形-树枝状嵌段共聚物(linear-dendriticblockcopolymers,ldbc),不仅结合了两类高分子的特点而具有独特的结构和性能,而且可通过对其树枝化基元和线形链的选择和结构修饰产生具有ph、温度、光、酶等环境刺激响应性的两亲性ldbc。酶响应性聚合物由于其良好的生物相容性,高灵敏性,精确的选择性,高效的酶催化效率以及温和的反应条件(溶液、ph=5-8、37℃)等优点,在生物材料和药物递送领域中具有潜在的应用价值。目前,关于对ph、温度、光等非特异性刺激信号有响应的ldbc已有较多的文献报道,而对酶有响应的ldbc仅见两篇文献报道,这两篇文献报道的酶响应性ldbc是采用先线形链法合成以不可生物降解的聚(乙二醇)(peg)为线形链、树枝化基元仅为二代,酶响应位点为树枝化基元上的4个可被青霉素g酰胺酶酶解的苯乙酰胺键。
目前,合成线形-树枝状嵌段共聚物的方法主要有三种,即先线形链(chain-first)法、先树枝法(dendron-first)和偶联(coupling)法。先线形链(chain-first)法也称为发散法,是先合成带一个活性端基的线形聚合物,然后通过发散增长树枝化基元;该方法具有高代数产物不易合成和纯化、结构有缺陷而且表征不易等缺点。先树枝法(dendron-first)也称为收敛法,是先合成带中心官能团的树枝化基元作为大分子引发剂,然后引发单体的聚合;用此法合成的线形-树枝状嵌段共聚物结构明确、易于表征,可以得到较高代数的产物。偶联(coupling)法是先分别合成线形链段和树枝化基元,然后使线形链的端基与树枝化基元的中心官能团进行偶合;这种方法的优点是可以得到一系列具有预设组成的线形-树枝状嵌段共聚物,然而,该方法需要选择一个有效的偶联反应。
两亲性ldbcs可以在选择性溶剂中自组装成不同形态的纳米结构,对疏水性小分子药物具有优异的负载能力,因此在药物控制释放和靶向释放方面具有较好的应用价值。目前,大多数两亲性线形-树枝状嵌段共聚物是以聚(乙二醇)(peg)为线形链,因为peg的生物相容性和低毒性。然而在过去几年中的研究发现,由于peg的不可生物降解性、热不稳定性、药代动力学行为的不可预期变化和静脉注射及口服后的过敏反应等缺陷,导致使用peg和含peg的聚合物可能产生不利影响,因此开发可保留peg的优点并克服其缺点的替代聚合物很有必要。聚(羟乙基-l-谷氨酰胺)(pheg)是一种亲水性的多肽,不仅具有优异的生物相容性,而且可生物降解,在生物医药领域有重要应用前景。目前,尽管有一些基于pheg的线形嵌段共聚物的文献报道,但以pheg为线形链的线形-树枝状嵌段共聚物却鲜见报道。
技术实现要素:
本发明的目的是提供一种蛋白酶响应性线形-树枝状嵌段共聚物及其制备方法和用途。
本发明共聚物为以聚(羟乙基-l-谷氨酰胺)(pheg)为线形链、以第2代树枝状半胱胺(g-b-pheg)为树枝化基元的线形-树枝状嵌段共聚物,或者是以聚(羟乙基-l-谷氨酰胺)(pheg)为线形链、以基于2,2’-二羟甲基丙酸(bis-mpa)的第1~4代脂肪族树枝状聚酯(fmocnh-bis-mpa-gn-b-pheg)为树枝化基元的线形-树枝状嵌段共聚物。
本发明以聚(羟乙基-l-谷氨酰胺)(pheg)为线形链、以第2代树枝状半胱胺(g-b-pheg)为树枝化基元的线形-树枝状嵌段共聚物,取名为g-b-pheg,其分子结构如下:
本发明以聚(羟乙基-l-谷氨酰胺)(pheg)为线形链、以基于2,2’-二羟甲基丙酸(bis-mpa)的第1~4代脂肪族树枝状聚酯(fmocnh-bis-mpa-gn-b-pheg)为树枝化基元的线形-树枝状嵌段共聚物,取名为fmocnh-bis-mpa-gn-b-pheg,其分子结构如下:
n为1~4的正整数。
本发明的g-b-pheg按以下步骤制备:
1、制备3,5-二(丙-2-炔基-1-氧)苯甲酸甲酯
按现有技术(例如可参照文献雷志丹,雷琳,李龙等.广东化工,2015,42(8):83-91),以碳酸钾为催化剂,丙酮为溶剂,用3,5-二羟基苯甲酸甲酯与3-溴丙炔反应,制备3,5-二(丙-2-炔基-1-氧)苯甲酸甲酯。
2、制备2-(叔丁氧羰基氨基)乙硫醇
按现有技术(例如可参照文献kimyz,kimjp.syntheticcommunications,2002,32(10):1601-1605),用2-氨基乙硫醇与二碳酸二叔丁酯反应制备2-(叔丁氧羰基氨基)乙硫醇。
3、制备树枝化基元(g-cooch3)
将步骤1得到的3,5-二(丙-2-炔基-1-氧)苯甲酸甲酯溶解在无水n,n-二甲基甲酰胺(dmf)中,每毫升dmf中溶有15~30mg的3,5-二(丙-2-炔基-1-氧)苯甲酸甲酯,加入步骤2得到的2-(叔丁氧羰基氨基)乙硫醇和偶氮二异丁腈(aibn),3,5-二(丙-2-炔基-1-氧)苯甲酸甲酯、2-(叔丁氧羰基氨基)乙硫醇和偶氮二异丁腈(aibn)的质量比为1.5~3:7~9:1,在氮气保护下,70~90℃反应10~14h,减压除去溶剂dmf,以石油醚与丙酮体积比为3:1的混合溶剂为洗脱剂进行柱色谱分离,制得树枝化基元(g-cooch3)。g-cooch3的分子结构如下:
4、制备树枝化基元引发剂(g-nh2)
在-5~5℃搅拌下,将步骤3得到的g-cooch3的甲醇溶液在0.8~1.2小时内逐滴加入到乙二胺的甲醇溶液中,每毫升甲醇中溶有70~90mgg-cooch3,每毫升甲醇中溶有100~120mg0.1~0.2ml乙二胺,g-cooch3和乙二胺的质量比为2~3:1,将上述混合物升至室温并于黑暗中搅拌90~100h。旋蒸除去甲醇得到粗产物,使用甲苯和甲醇体积比为9:1)的共沸混合物除去过量的乙二胺后得到树枝化基元引发剂(g-nh2)。g-nh2的分子结构如下:
5、制备l-谷氨酸苄酯-n-羧酸酐(blg-nca)
按现有技术(例如可参照文献dalywh,pochéd.tetrahedronletters,1988,29(46):5859-5862),用l-谷氨酸苄酯与三聚光气反应制备l-谷氨酸苄酯-n-羧酸酐(blg-nca)。
6、制备线形-树枝状嵌段共聚物(g-b-pblg)
将步骤5得到的l-谷氨酸苄酯-n-羧酸酐(blg-nca)用无水dmf溶解后,加入步骤4得到的g-nh2的无水dmf溶液,每毫升dmf中溶有blg-nca380~420mg,每毫升dmf中溶有g-nh2100~200mg,blg-nca与g-nh2的质量比为3.5~4.5:1,将上述反应物通氮气、于25~35℃反应1.5~2.5天,然后用无水乙醚沉淀2~4次,真空干燥18~28h,得到线形-树枝状嵌段共聚物(g-b-pblg)。g-b-pblg的分子结构如下:
7、制备线形-树枝状嵌段共聚物(g-b-pheg)
将步骤6得到的g-b-pblg和2-羟基吡啶溶解在dmf中,g-b-pblg和2-羟基吡啶的质量比为2~4:1,每毫升dmf中溶有100~130mg的g-b-pblg,然后逐滴加入乙醇胺,乙醇胺和g-b-pblg的质量比为6~10:1,上述混合物在氮气保护下35~45℃搅拌20~28h,将反应液用乙醚共沉淀后,溶于适量蒸馏水中,透析3~5天,冷冻干燥得到g-b-pheg。
上述步骤中的过滤,旋蒸,柱色谱,真空干燥等工序同常规技术,
本发明的fmocnh-bis-mpa-gn-b-pheg按以下步骤制备:
1、制备异亚丙基-2,2-二(甲氧基)丙酸
按现有技术(例如可参照文献movellanj,urbanp,molese,et,al.biomaterials,2014,35(27):7940-7950),用2,2-二(羟甲基)丙酸与2,2-二甲氧基丙烷反应制备异亚丙基-2,2-二(甲氧基)丙酸。
2、制备异亚丙基-2,2-二(甲氧基)丙酸酐
按现有技术(例如可参照文献movellanj,urbanp,molese,et,al.biomaterials,2014,35(27):7940-7950),用步骤1得到的异亚丙基-2,2-二(甲氧基)丙酸为原料,以n,n'-二环己基碳二亚胺为脱水剂制备异亚丙基-2,2-二(甲氧基)丙酸酐。
3、制备boc单保护乙二胺-g1-缩酮
将步骤3得到的异亚丙基-2,2-二(甲氧基)丙酸和n,n'-二环己基碳二亚胺(dcc)溶于二氯甲烷,异亚丙基-2,2-二(甲氧基)丙酸和dcc的质量比为10~15:1,每毫升二氯甲烷中溶有50~80mg异亚丙基-2,2-二(甲氧基)丙酸;冷至-5~5℃,在氮气保护下,滴加溶有n-叔丁氧羰基乙二胺(boc单保护乙二胺)和4-二甲氨基吡啶(dmap)的二氯甲烷溶液,boc单保护乙二胺和4-二甲氨基吡啶(dmap)的质量比为1:1~1.5,每毫升二氯甲烷中溶有80~100mg的boc单保护乙二胺;滴加完毕后,室温下搅拌20~30h,过滤,旋蒸除去溶剂后,粗产物以石油醚与丙酮体积比为10~5:1的混合溶剂为洗脱剂进行硅胶柱色谱分离得到boc单保护乙二胺-g1-缩酮。boc单保护乙二胺-g1-缩酮的分子结构如下:
4、制备第1代基于2,2’-二羟甲基丙酰胺(bis-mpa)的树枝化基元bis-mpa-g1-nhboc
在溶有步骤3得到的boc单保护乙二胺-g1-缩酮的甲醇溶液中,加入对甲苯磺酸一水合物,每毫升甲醇中溶有80~120mgboc单保护乙二胺-g1-缩酮,boc单保护乙二胺-g1-缩酮和对甲苯磺酸一水合物的质量比为7~9:1。室温搅拌12~18h后,加入适量饱和nahco3水溶液猝灭,乙酸乙酯萃取,经无水mgso4干燥,过滤,旋蒸溶剂,得到树枝化基元bis-mpa-g1-nhboc。bis-mpa-g1-nhboc的分子结构如下:
5、制备第1代树枝化基元fmocnh-bis-mpa-g1-nhboc
将步骤4得到的bis-mpa-g1-nhboc溶解在二氯甲烷(ch2cl2)和dmf的混合溶剂中,二氯甲烷(ch2cl2)和dmf体积比为4~6:1,每毫升二氯甲烷和dmf的混合溶剂中溶有15~25mg的bis-mpa-g1-nhboc,加入n-芴甲氧羰基-甘氨酸(fmoc-甘氨酸)和4-二甲氨基吡啶,其中ho-g1-nhboc、fmoc-甘氨酸和4-二甲氨基吡啶的质量比为1~2:3.5~5.5:1。搅拌溶解完全后,冷却至-5~5℃,在氮气保护下逐滴加入溶有n,n'-二环己基碳二亚胺(dcc)的二氯甲烷溶液,每毫升二氯甲烷中溶有200~260mg的dcc;滴加完毕后,继续在-5~5℃反应0.5~1.5h后,室温下反应20~28h,过滤,旋蒸溶剂,以石油醚与丙酮体积比为1~3:1的混合溶剂为洗脱剂进行柱色谱分离,得到fmocnh-bis-mpa-g1-nhboc。fmocnh-bis-mpa-g1-nhboc的分子结构如下:
6、制备第1代树枝化基元引发剂fmocnh-bis-mpa-g1-nh2
在冰水浴下,将步骤5制备的bis-mpa-g1-nhboc溶解在三氟乙酸(tfa)的ch2cl2溶液中,其中每毫升三氟乙酸(tfa)的ch2cl2溶液中溶有10~20mg的g1-nhboc,三氟乙酸(tfa)与ch2cl2的体积比为1:0.5~1.5;搅拌反应5~7h后,加入饱和nahco3溶液调至ph=8-9,经ch2cl2萃取、无水mgso4干燥、过滤、浓缩后得到fmocnh-bis-mpa-g1-nh2。fmocnh-bis-mpa-g1-nh2的分子结构如下:
7、制备第1代线形-树枝状嵌段共聚物fmocnh-bis-mpa-g1-b-pblg
将制备g-b-pheg步骤5中制备的blg-nca用无水dmf溶解,每毫升dmf中溶有400~460mg的blg-nca;加入溶有步骤6制备的fmocnh-bis-mpa-g1-nh2的无水dmf溶液,每毫升dmf中溶有100~200mg的fmocnh-bis-mpa-g1-nh2,blg-nca与fmocnh-bis-mpa-g1-nh2的质量比为10~20:1;在氮气保护下28~35℃下反应1.5~2.0天,然后用无水乙醚沉淀2~5次,真空干燥20~30h,得到fmocnh-bis-mpa-g1-b-pblg。fmocnh-bis-mpa-g1-b-pblg的分子结构如下:
8、制备第1代聚[n-(2-羟乙基-l-谷氨酰胺)]-b-树枝状聚酯fmocnh-bis-mpa-g1-b-pheg
将步骤7得到的fmocnh-bis-mpa-g1-b-pblg和2-羟基吡啶溶解在dmf中,然后逐滴加入乙醇胺,其中fmocnh-bis-mpa-g1-b-pblg、2-羟基吡啶和乙醇胺的质量比为1.5~3:1:16~30,每毫升dmf中溶有100~160mg的fmocnh-bis-mpa-g1-b-pblg;氮气保护下30~50℃搅拌反应20~30h,将反应液用乙醚共沉淀后,溶于适量蒸馏水中,透析2~5天,冷冻干燥得到fmocnh-bis-mpa-g1-b-pheg。fmocnh-bis-mpa-g1-b-pheg的分子结构如下:
9、制备boc单保护乙二胺-g2-缩酮
将步骤4制备的bis-mpa-g1-nhboc、步骤2制备的异亚丙基-2,2-二(甲氧基)丙酸酐、4-二甲氨基吡啶溶解在吡啶和ch2cl2的混合溶液中,其中bis-mpa-g1-nhboc、异亚丙基-2,2-二(甲氧基)丙酸酐与4-二甲氨基吡啶的质量比为0.5~1.5:3~5:1,吡啶与ch2cl2的体积比为1~2:1,每毫升吡啶和ch2cl2的混合溶液中溶有60~100mg的bis-mpa-g1-nhboc,室温反应10~20h。加入8~15ml吡啶和水混合溶液(体积比1:1~2)猝灭除去过量的酸酐,有机相加入80~120mlch2cl2稀释,并依次用10%nahso4、10%na2co3、饱和nacl溶液萃取2~5次,每次每种溶液40~60ml,有机相用无水mgso4干燥,过滤,旋蒸溶剂,以石油醚与丙酮体积比为4~8:1的混合溶剂为洗脱剂进行柱色谱分离,得到boc单保护乙二胺-g2-缩酮。boc单保护乙二胺-g2-缩酮的分子结构如下:
10、制备第2代基于2,2’-二羟甲基丙酰胺(bis-mpa)的树枝化基元bis-mpa-g2-nhboc
在溶有步骤9制备的boc单保护乙二胺-g2-缩酮的甲醇溶液中,加入强酸性阳离子交换树脂,每毫升甲醇中溶有100~200mg的boc单保护乙二胺-g2-缩酮,boc单保护乙二胺-g2-缩酮与强酸性阳离子交换树脂的质量比为1:1.5~2.5;室温反应6~10h后,过滤除去强酸性阳离子交换树脂后,浓缩滤液得到bis-mpa-g2-nhboc。bis-mpa-g2-nhboc的分子结构如下:
11、制备第2代树枝化基元fmocnh-bis-mpa-g2-nhboc
将步骤10制备的bis-mpa-g2-nhboc溶解在ch2cl2和dmf混合溶剂中,加入fmoc-甘氨酸,其中每毫升ch2cl2和dmf混合溶剂中溶有2~6mg的bis-mpa-g2-nhboc,bis-mpa-g2-nhboc与fmoc-甘氨酸的质量比为1:2.5~5,ch2cl2和dmf的体积比为4~6:1;搅拌至完全溶解后,加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺(edc)和dmap,bis-mpa-g2-nhboc、edc与dmap的质量比为1~1.5:3~6:1;氮气保护下搅拌反应10~16h,旋蒸除去溶剂,残余物溶于乙酸乙酯,依次用1~1.5mkhso4、1~1.5mnaco3、饱和食盐水洗涤2~5次,每次每种溶液40~60ml。有机相用mgso4干燥、过滤、浓缩得到fmocnh-bis-mpa-g2-nhboc。fmocnh-bis-mpa-g2-nhboc的分子结构如下:
12、制备第2代树枝化基元引发剂fmocnh-bis-mpa-g2-nh2
以步骤11得到的fmocnh-bis-mpa-g2-nhboc为原料,重复步骤6制得fmoc-bis-mpa-g2-nh2。fmocnh-bis-mpa-g2-nh2的分子结构如下:
13、制备第2代树枝状聚酯-b-聚[n-(2-羟乙基-l-谷氨酰胺)]fmocnh-bis-mpa-g2-b-pheg
以步骤12得到的fmoc-bis-mpa-g2-nh2为引发剂,重复步骤7~8制得fmoc-bis-mpa-g2-b-pheg。fmocnh-bis-mpa-g2-b-pheg的分子结构如下:
14、制备第3代和第4代树枝状聚酯-b-聚[n-(2-羟乙基-l-谷氨酰胺)]fmocnh-bis-mpa-g3-b-pheg和fmocnhbis-mpa-g4-b-pheg
以步骤10得到的fmocnh-bis-mpa-g2-nhboc为原料,重复步骤11~13,得到fmocnh-bis-mpa-g3-b-pheg;再以此过程中得到的fmocnh-bis-mpa-g3-nhboc为原料,重复步骤11~13,得到fmocnh-bis-mpa-g4-b-pheg。fmocnh-bis-mpa-g3-b-pheg和fmocnh-bis-mpa-g4-b-pheg的分子结构如下:
上述步骤中的过滤,旋蒸,柱色谱,真空干燥等工序同常规技术,
以上步骤可以是进行到制备出第1代的目的产物后就结束,也可以是制备出大于第1代的、相应代数的目的产物后才结束。
本发明中,所制备的树枝状半胱胺-b-聚[n-(2-羟乙基-l-谷氨酰胺)](g-b-pheg)和树枝状聚酯-b-聚[n-(2-羟乙基-l-谷氨酰胺)](fmocnh-bis-mpa-gn-b-pheg)结构明确,都具有较窄的分子量分布(pdi<1.2),都可以通过1hnmr、gpc和maldi-tofms进行详细表征。
本发明制备的蛋白酶响应性线形-树枝状嵌段共聚物的用途是可以在水溶液中自组装为核壳结构胶束,用于包载难溶性药物或亲脂性的染料。最好是在蛋白酶的作用下释放出所包载得药物或染料。
本发明的有益效果:本发明制备的线形-树枝状嵌段共聚物具有两亲性,在水溶液中能自组装为以亲脂性的树枝化基元为核、以亲水性的聚[n-(2-羟乙基-l-谷氨酰胺)]为壳的胶束,并可以包载亲脂性的染料、抗癌药物阿霉素、紫杉醇等,在木瓜蛋白酶或组织蛋白酶b(cathepsinb)作用下,胶束解组装释放出包载的亲脂性染料或药物,组织蛋白酶b(cathepsinb)是一种在肿瘤细胞中高表达的酶,因此,本发明研制的线形-树枝状嵌段共聚物可望成为较为理想的难溶性抗癌药物载体,用于抗癌药物的靶向释放。与文献报道的经典以peg为线形链的线形-树枝状嵌段共聚物相比,本发明合成的以聚(羟乙基-l-谷氨酰胺)(pheg)为线形链的线形-树枝状杂化嵌段共聚物是可以生物降解的;而且与文献报道的酶响应性线形-树枝状杂化嵌段共聚物相比,本发明制备的以聚(羟乙基-l-谷氨酰胺)(pheg)为线形链的线形-树枝状杂化嵌段共聚物整个线形链可以被在肿瘤细胞中高表达的组织蛋白酶b降解成无毒的氨基酸,酶响应位点多,共聚物胶束解组装较快,因此本发明制备的线形-树枝状杂化嵌段共聚物更适合于抗癌药物的靶向释放。本发明制备的线形-树枝状嵌段共聚物带有强亲脂性fmoc端基,使其自组装形成的胶束更稳定。本发明采用“先树枝(dendron-first)法”合成以聚(羟乙基-l-谷氨酰胺)(pheg)为线形链的线形-树枝状杂化嵌段共聚物,结构明确且易于表征,结构无缺陷,可以得到高代数的酶响应性线形-树枝状杂化嵌段共聚物。
附图说明
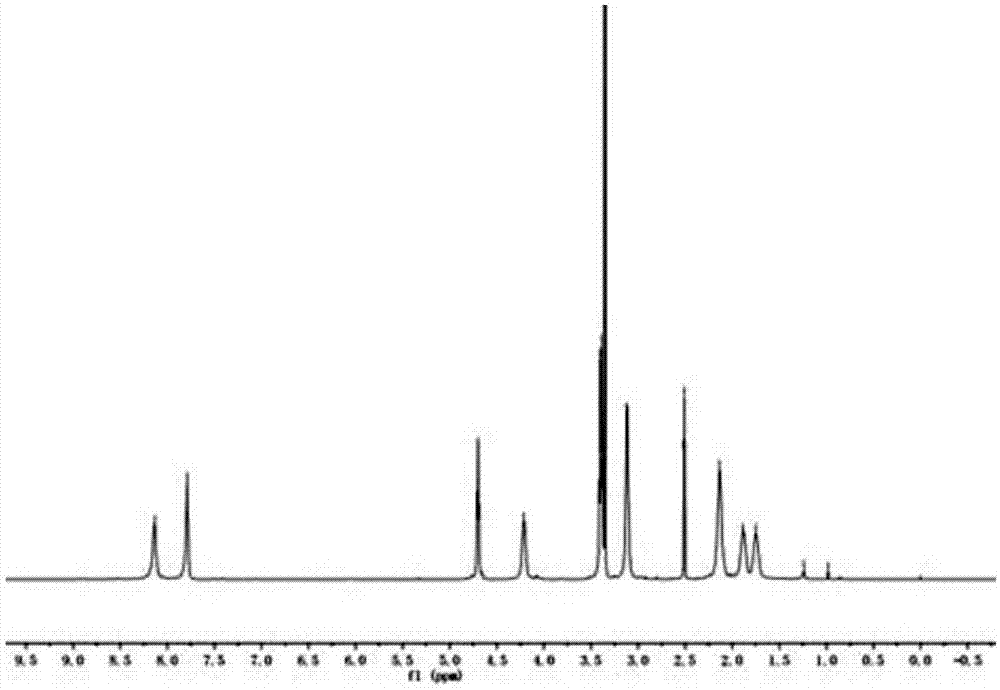
图1是实施例二十一中得到的fmocnh-bis-mpa-g2-pheg的核磁共振氢谱。
图2实施例二十三中加入木瓜蛋白酶前(a)后(b)g-b-pheg胶束透射电镜(tem)图。
图3是实施例二十三中包载尼罗红的g-b-pheg胶束(酶浓度为0μm)和加入木瓜蛋白酶(酶浓度为80μm)的荧光强度(纵坐标)随时间(横坐标)的变化图。
具体实施方式
下面用实施例来进一步详述本发明,但本发明的内容并不局限于此。
实施例一:
将5g的3,5-二羟基苯甲酸甲酯、5ml溴丙炔、12.3g无水碳酸钾、100ml丙酮加到圆底烧瓶中,室温搅拌16h后,过滤,旋蒸溶剂,将反应物溶于乙酸乙酯,用蒸馏水洗涤三次,干燥、过滤、浓缩有机相得到粗产物。用石油醚与乙酸乙酯体积比为5:1的混合溶剂为洗脱剂进行柱色谱分离,得到3,5-二(丙-2-炔基-1-氧)苯甲酸甲酯,产率为60.7%。
实施例二:
将1g的2-氨基乙硫醇盐酸盐溶解在10ml无水四氢呋喃中,在0℃搅拌下,加入0.53g的nah,5min后升温至室温,搅拌10min。接着将反应混合物再次冷却到0℃,加入2.29g的boc2o。20min后,移去冰浴,在室温下继续搅拌2h,然后用饱和碳酸氢钠溶液猝灭,将混合物倒入蒸馏水中,并用乙酸乙酯萃取三次,有机层合并后用无水mgso4干燥,过滤、旋蒸溶剂,得粗产物。以石油醚与乙酸乙酯体积比为3:1的混合溶剂为洗脱剂进行柱色谱分离,得到2-(叔丁氧羰基氨基)乙硫醇,产率为89%。
实施例三:
在250ml的烧瓶中,将0.65g的3,5-二(丙-2-炔基-1-氧)苯甲酸甲酯溶解在30ml无水dmf溶液中,加入2.7ml的2-(叔丁氧羰基氨基)乙硫醇、350mg的aibn,升温至80℃于氮气保护下反应12h,减压除去溶剂dmf,以石油醚与丙酮体积比为3:1的混合溶剂为洗脱剂进行柱色谱分离,最后得到树枝化基元g-cooch3,产率为83.5%。
实施例四:
0℃搅拌下,将10ml溶有0.82g的g-cooch3的甲醇溶液逐滴加入到20ml含2.63ml乙二胺的甲醇溶液中,然后将混合物升至室温搅拌96h。旋蒸除去甲醇得到粗产物,使用甲苯和甲醇(9:1v/v)的共沸混合物(共沸点63.5℃)移除过量的乙二胺后得到树枝化基元引发剂(g-nh2),产率为84.1%。
实施例五:
氮气保护下在10gl-谷氨酸苄酯中加入100ml无水thf,升温至50℃,搅拌下加入10g三聚光气,反应30mins后,倒入400ml石油醚中,放入冰箱中过夜。过滤,将粗品用乙酸乙酯溶解后,分别用冷的饱和nahco3溶液和nacl溶液洗涤,有机相加入无水mgso4干燥。过滤,滤液倒入石油醚充分沉淀。粗产物再用乙酸乙酯、石油醚重结晶得到blg-nca,产率48%。
实施例六:
将2.01g的blg-nca单体用5ml无水dmf溶解,再加入3ml溶有0.5g的g-nh2的无水dmf溶液,氮气保护下30℃反应2天,然后用无水乙醚沉淀3次,真空干燥24h,得到g-b-pblg,产率76.4%。
实施例七:
将1.1g的g-b-pblg和0.5g的2-羟基吡啶溶解在10mldmf中,然后逐滴加入8ml乙醇胺,于40℃氮气下搅拌24h,将反应液用乙醚共沉淀后,溶于适量蒸馏水中,透析3天,冷冻干燥得到g-b-pheg,产率70%。
实施例八:
在250ml烧瓶中加入150ml丙酮、30.1g二羟甲基丙酸、41.4ml的2,2-二甲氧基丙烷、2.3g对甲苯磺酸一水合物,室温反应2h后,加入3ml氨水/乙醇混合液(4:1v/v)中和,蒸发丙酮,残留物溶解在适量乙酸乙酯中,蒸馏水萃取三次,有机相经无水mgso4干燥,过滤,旋蒸溶剂,得到26.11g异亚丙基-2,2-二(甲氧基)丙酸,产率为68.2%。
实施例九:
将10g的异亚丙基-2,2-二(甲氧基)丙酸溶解在30mlch2cl2中,并加入10ml溶有5.9gn,n'-二环己基碳二亚胺(dcc)的ch2cl2溶液。室温下搅拌48h,过滤,用少量ch2cl2洗涤,旋蒸溶剂,所得残余物用200ml正己烷稀释,剧烈搅拌至白色浑浊,放置冰箱冷藏过夜,过滤得到异亚丙基-2,2-二(甲氧基)丙酸酐,产率为66%。
实施例十:
n2保护下,将5g异亚丙基-2,2-二(甲氧基)丙酸、5.9g的dcc溶于80ml的ch2cl2。0℃,缓慢滴加溶有4.6g单boc保护的乙二胺、355mgdmap的50mlch2cl2溶液,滴完,继续在0℃搅拌1h,升至室温继续反应24h。过滤,旋蒸溶剂,以石油醚与丙酮体积比为6:1的混合溶剂为洗脱剂进行柱色谱分离得到boc单保护乙二胺-g1-缩酮。产率为77.2%。
实施例十一:
在100ml溶有10g的boc单保护乙二胺-g1-缩酮的甲醇溶液中,加入1.2g对甲苯磺酸一水合物。室温搅拌15h后,加入适量饱和nahco3水溶液猝灭,乙酸乙酯萃取,经无水mgso4干燥,过滤,旋蒸溶剂,得到bis-mpa-g1-nhboc,产率为91.6%。
实施例十二:
将1g的bis-mpa-g1-nhboc溶解在48mlch2cl2和dmf混合溶液(5:1v/v)中,并加入3.23g的fmoc-甘氨酸,0.66g的dmap。原料完全溶解后,将体系冷却至0℃,并于n2保护下逐滴加入溶有2.24g的dcc的二氯甲烷溶液。滴完,继续在0℃反应1h,升至室温、于氮气保护下反应24h,过滤,旋蒸溶剂,以石油醚与丙酮体积比为2:1的混合溶剂为洗脱剂进行柱色谱分离,得到fmocnh-bis-mpa-g1-nhboc,产率为74.2%。
实施例十三:
0℃,将0.48g的fmocnh-bis-mpa-g1-nhboc溶解在30ml50%的tfa/ch2cl2混合溶液中,搅拌6h后,加入饱和nahco3调ph=8-9,经ch2cl2萃取、无水mgso4干燥、过滤、浓缩得到fmocnh-bis-mpa-g1-nh2,产率为78.8%。
实施例十四:
将4.3g的blg-nca单体用10ml无水dmf溶解,加入2ml溶有0.3g的fmocnh-bis-mpa-g1-nh2的无水dmf溶液,氮气保护下30℃反应2天,粗产物用无水乙醚沉淀三次,真空干燥24h,得到fmocnh-bis-mpa-g1-b-pblg。
实施例十五:
将2.6g的fmocnh-bis-mpa-g1-b-pblg和1.1g的2-羟基吡啶溶解在20mldmf中,然后逐滴加入20ml乙醇胺,氮气保护下40℃搅拌24h,将反应液用乙醚共沉淀后,溶于适量蒸馏水中,透析3天,冷冻干燥得到fmocnh-bis-mpa-g1-b-pheg,产率为78.4%。
实施例十六:
将6g的bis-mpa-g1-nhboc、21.6g异亚丙基-2,2-二(甲氧基)丙酸酐、5.4g的dmap溶解在80ml吡啶和ch2cl2混合溶液(3:2v/v)中,室温反应18h。加入10ml吡啶和水混合溶液(1:1v/v)猝灭反应,在有机相中加入100mlch2cl2稀释,并依次用10%nahso4(3×50ml)、10%na2co3(3×50ml)、nacl(50ml)萃取,有机相用无水mgso4干燥,过滤,旋蒸溶剂,以石油醚与丙酮体积比为6:1的混合溶剂为洗脱剂进行柱色谱分离,得到boc单保护乙二胺-g2-缩酮,产率为72.54%。
实施例十七:
在50ml溶有7.54g的boc单保护乙二胺-g2-缩酮的甲醇溶液中,加入15g强酸性阳离子交换树脂。室温反应8h后,过滤、浓缩滤液得到bis-mpa-g2-nhboc,产率为95.7%。
实施例十八:
将0.2g的bis-mpa-g2-nhboc溶解在48mlch2cl2和dmf混合溶液(5:1v/v)中,并加入0.71g的fmoc-甘氨酸,搅拌至完全溶解后,加入0.69g的edc和0.15g的dmap。氮气保护下搅拌反应12h,旋蒸溶剂,残余物溶于乙酸乙酯,依次用1mkhso4(3×50ml),1mnaco3(3×50ml),饱和食盐水洗涤。有机相用mgso4干燥、过滤、浓缩得到fmocnh-bis-mpa-g2-nhboc。
实施例十九:
冰水浴下,将2.7g的fmocnh-bis-mpa-g2-nhboc溶解在30ml50%的tfa/ch2cl2混合溶液中,搅拌6h后,加入饱和nahco3调ph=8-9,经ch2cl2萃取、无水mgso4干燥、过滤、浓缩得到fmocnh-bis-mpa-g2-nh2,产率为71.1%。
实施例二十:
将3.16g的blg-nca用8ml无水dmf溶解,再加入4ml溶有0.46g的fmocnh-bis-mpa-g2-nhboc的无水dmf溶液,在氮气保护下30℃反应2天,粗产物用无水乙醚沉淀三次,真空干燥24h,得到fmocnh-bis-mpa-g2-pblg,产率为64.4%。
实施例二十一:
将2.1g的fmocnh-bis-mpa-g2-pblg和0.9g的2-羟基吡啶溶解在18mldmf中,然后逐滴加入18ml乙醇胺,于40℃氮气保护下搅拌24h,粗产物用乙醚共沉淀后,溶于适量蒸馏水中,透析3天,冷冻干燥得到fmocnh-bis-mpa-g2-pheg,产率为65%。
实施例二十二:
以实施例十八得到的fmocnh-bis-mpa-g2-nhboc为原料重复实施例十九~实施例二十一的步骤得到fmocnh-bis-mpa-g3-pheg;重复相同的步骤得到fmocnh-bis-mpa-g4-pheg。
实施例二十三:
在250μm的g-b-pheg胶束缓冲溶液(pbs,ph=6.5)中加入250μm的尼罗红溶液(将尼罗红丙酮溶液加入到ph为6.5的pbs缓冲溶液中配制得到),室温搅拌6h,静置,离心分离未包载的尼罗红,收集上清液。在上清液中加入活化后的80μm木瓜蛋白酶pbs缓冲溶液(ph=6.5),37℃孵育,以未加入木瓜蛋白酶的g-b-pheg胶束包载尼罗红的溶液进行对照,进行荧光光谱分析、粒径测定和透射电镜(tem)观测。结果表明,g-b-pheg在水溶液中能自组装为球形纳米胶束,此胶束能包载难溶性的尼罗红染料,加入木瓜蛋白酶后,胶束解聚释放出包载的尼罗红。
实施例二十四:
将共聚物换成fmocnh-bis-mpa-gn-b-pheg(n=1~4),尼罗红换成阿霉素,重复实施例二十三的步骤制备fmocnh-bis-mpa-gn-b-pheg包载阿霉素胶束,加入组织蛋白酶b(cathepsinb),以未加酶的fmocnh-bis-mpa-gn-b-pheg胶束包载阿霉素的溶液进行对照,进行荧光光谱分析、粒径测定和透射电镜(tem)观测。结果表明,fmocnh-bis-mpa-gn-b-pheg在水溶液中能自组装为纳米胶束,此胶束能包载难溶性的抗癌药物阿霉素,加入组织蛋白酶b后,胶束解聚释放出包载的阿霉素。fmocnh-bis-mpa-gn-b-pheg的代数对药物的载药量、包封率和释放速率都有影响,通过调节线形-树枝状嵌段共聚物的代数,可以优化其胶束对药物的载药量、包封率和释放速率。
- 还没有人留言评论。精彩留言会获得点赞!
- OTUD5的单克隆抗体制备及其在癌症治疗中的用途的制造方法与工艺
- 一种胱氨酸蛋白酶抑制剂C测定试剂盒的制造方法与工艺
- 氮氧化物作为新型组蛋白去乙酰化酶抑制剂的抗肿瘤应用的制造方法与工艺
- 一种硼替佐米水溶性药用组合物及其制备方法和用途与制造工艺
- 初级癌症疗法后使用的含硼蛋白酶体抑制剂的制造方法与工艺
- 斯氏副柔线虫半胱氨酸蛋白酶抑制剂Pj_CPI基因克隆、重组表达方法及应用与制造工艺
- 一种制备自噬小体型瘤苗的培养基添加剂及其制备方法与制造工艺
- 一种高产脯氨酸蛋白酶的黑曲霉菌株的制造方法与工艺
- N-萘烷基取代的含氮杂环衍生物、制备方法及其抗肿瘤用途与制造工艺
- 作为BET蛋白抑制剂的1H‑吡咯并[2,3‑c]吡啶‑7(6H)‑酮和吡唑并[3,4‑c]吡啶‑7(6H)‑酮的制造方法与工艺