检测IL-28B基因单核苷酸多态性的荧光原位杂交测序方法与流程
本发明公开了一种单核苷酸多态性的检测方法,属于分子遗传学领域。
背景技术:
随着医药科学发展的不断进步,全世界每年有很多的药物出现,因不良反应太大而在药物研究中被淘汰。虽然药物的效果和不良反应与人种及个体差异有关,但人与人之间基因组序列的遗传差异是药物疗效和不良反应的重要原因。单核苷酸多态性(singlenucleotidepolymorphisms;snp)就是人与人之间1个碱基之间的差异,基因组序列差异有好几种,snp仅是最常见最普通的一种,在临床上意义重大。
snp是由单个碱基改变而引起的dna序列的多态性。包括单碱基的转换,颠倒以及单碱基的插入或者缺失等。snp在人基因组中的发生频率比较高,大约平均每1000个碱基中就有一个多态性位点,是继微卫星之后的第三代遗传标记,有些snp位点还会影响基因的功能,从而导致生物性状改变甚至致病。snp是人类可遗传的变异种最常见的一种,占所有已知多态性的90%以上,目前dbsnp已经收录了约500万个已验证的人类snp数据。
在遗传学分析中,snp作为一类遗传标记得以广泛应用,主要源于这几个特点:
(1)密度高snp在人类基因组的平均密度估计为1/1000bp,在整个基因组的分布达3×106个,遗传距离为2~3cm,密度比微卫星标记更高,可以在任何一个待研究基因的内部或附近提供一系列标记;
(2)富有代表性某些位于基因内部的snp有可能直接影响蛋白质结构或表达水平,因此,它们可能代表疾病遗传机理中的某些作用因素;
(3)遗传稳定性与微卫星等重复序列多态性标记相比,snp具有更高的遗传稳定性。
(4)易实现分析的自动化snp标记在人群中只有两种等位型(allele)。这样在检测时只需一个“+\-”或“全\无”的方式,而无须像检测限制性片段长度多态性,微卫星那样对片段的长度做出测量,这使得基于snp的检测分析方法易实现自动化。
snp不仅可以为药物研发的源头上提供重要的靶标信息,也能够为治疗终端的个体化给药方案提供指导意见,以最大限度提供药物疗效和降低药物应用中产生的毒副反应,其重要意义体现于:
(1)提高新药筛选的准确性:最近研究表明,通过测定不同人群组之间的snp,可有效地寻找新药的目标分子。由于患者间的snp差异,snp的研究结果可以用于降低已知药物和临床试验药物的不良反应,并提高其疗效。也可以挽救因不良反应大而被淘汰的优良药物,只要找出与其相关联的snp就可以作为有效药物用于临床。因为药物之间相互影响或改变各自的代谢途径,从而产生协同作用而提高药效,也可根据snp选择患者进行临床试验,提高临床研究质量,增强新药成功率,而且从疾患基因的snp出发,研究开发目标药物是很理想的。
(2)药物的应答性与snp:当给患有同种疾病的患者服用相同的药物时,不同的患者会有不同的反应,分为显著有效者,有效者,无效者。在临床治疗中,通常医师无法根据患者的症状来决定患者对什么药物最有效,只能依赖临床研究数据的平均统计结果和个人经验来选择。但是这种症状或疾病的患者,即使应用相同的药物也会产生不同的治疗结果,这种差异可通过检测snp来预测。如果snp正好位于表达的基因上,则产生的表达产物-蛋白的氨基酸序列有可能不同,由此就可能是药物作用的靶位发生改变,对药物的亲和力下降或者消失。所以对患者进行snp分析可达到正确的诊断和有效的治疗目的。
(3)snp与药物的不良反应:在医疗过程中,某些患者服用药物后不但无效,有的还会产生严重的不良反应,如处理不当有时还会引起死亡。据统计,中国每年5000万住院病人中至少250万人的入院治疗与药物的不良反应有关,其中50万人属于严重不良反应,因此致死的人数每年约为19.2万人,比传染病致死的人数高出数倍,另外有调查,中国的180万聋哑儿童中,有60%以上是由于不合理用药所导致的。在治疗前了解患者的snp,建立健康人和患者的snp数据库,并进行相关性分析十分重要。
干扰素是人体免疫系统中的淋巴细胞在病毒刺激下产生的一种淋巴因子,具有一定的抗病毒作用。人体内的干扰素有许多亚型。最初被发现的干扰素(interleukin)有α、β、γ,干扰素α和β又被称为ⅰ型干扰素,干扰素γ为ⅱ型。2003年,美国科学家发现了人体内还有第ⅲ型干扰素,也称为λ干扰素。λ干扰素还分为λ-1干扰素。λ-2干扰素和λ-3干扰素,其中λ-3干扰素(ifn-λ3)不仅具有明确的抗病毒作用,还能促进α干扰素的产生。
深入研究的结果发现,λ-3干扰素是白细胞介素28b的基因编码的一种细胞因子,简称其为“il28b”。人类的“il28b基因”表型主要在3种类型:c/c型、c/t型和t/t型。人们发现,“il28b基因”为c/c型者,可能有利于丙肝病毒感染后的自发性清除,对干扰素治疗的应答也比c/t型和t/t型好。il-28b基因的多态性和种族分布有关系,我国的丙肝病毒感染者中il28b等位基因以c/c型为主。
il-28b*1(rs12979860)位于ifn-λ3编码区序列上游3000个碱基对处的内含子上,其基因表型有c/c,c/t和t/t3种,其中亚州东部人群中,cc的基因型概率最高。il-28b*2(rs8099917)是il-28b上游8kb的一个snp位点,该基因型分为gg,gt,tt基因型,其中基因型为gg的患者对标准疗法的失败率是gt或者tt基因型患者的2倍。il-28b*3(rs12980275)位于il-28b基因下游24000个碱基对处,也可用于预测1型hcv患者接受标准治疗方案的治疗疗效,包括a/a、a/g和g/g型,其中a/a为保护性基因型,rs12980275a/a型与我国汉族人群中hcv自发清除情况相关。il-28b*1(rs12979860)是il-28b区域附近7个snp位点与治疗应答有明显相关性的与治疗应答相关性最强的一个。临床数据表示,il-28b基因多态性与患者对聚乙二醇化干扰素治疗具有的持续病毒学反应有关。il-28*1cc型的病毒治疗完全有效率,是tt型的3倍;il-28*2gg型患者,则对丙肝治疗完全无效。
药物基因组学的高速发展为个体化给药提供了基础,而真正实现个体化给药,基因多态性检测成为关键。目前已有多种方法可用于snp检测,传统的方法是采用一些已有的成熟技术,如dna双脱氧测序、dna焦磷酸测序、全基因组测序、单链构象多态性(sscp)、限制性酶切片段长度多态性(rflp)、变性梯度凝胶电泳(dgge)等。新的高通量测定snp的方法有芯片技术、变性高效液相色谱(dhplc)、动态等位基因特异的杂交、基质辅助激光解析/电离-飞行时间质朴(maldi-tofms)技术等。
(1)dna双脱氧测序、焦磷酸测序、全基因组测序:通过对不同个体同一基因或基因片段进行测序和序列比较,以确定所研究的碱基是否变异,其检出率可达100%。虽然可以得到snp的类型及其准确位置等snp分型所需要的重要参数。但是此方法操作繁琐,成本较高,不适用于普遍操作。
(2)sscp:单链dna在中性条件下会形成二级结构,不同的二级结构在电泳中会出现不同的迁移率。这种二级结构依赖于碱基的组成,单个碱基的改变也会影响其构象,最终会导致在凝胶上迁移速度的改变。在非变性聚丙烯酰胺凝胶上,短的单链dna和rna分子依其单碱基序列的不同而形成不同的构象,这样在凝胶上的迁移速率不同,出现不同的条带,检测snp。这种方法的弊端在于不能确定突变类型和具体位置。
(3)rflp:利用限制性内切酶的酶切位点的特异性,用两种或两种以上的限制性内切酶作用于同一dna片断,如果存在snp位点,酶切片断的长度和数量则会出现差异,根据电泳的结果就可以判断是否snp位点。但是该技术应用的前提是snp的位点必须含有该限制内切酶的识别位点。
(4)dgge:是利用长度相同的双链dna片段解链温度不同的原理,通过梯度变性胶将dna片段分开的电泳技术。电泳开始时,dna在胶中的迁移速率仅与分子大小有关,而一旦dna泳动到某一点时,即到达该dna变性浓度位置时,使得dna双链开始分开,从而大大降低了迁移速率。当迁移阻力与电场力平衡时,dna片段在凝胶中基本停止迁移。由于不同的dna片段的碱基组成有差异,使得其变性条件产生差异,从而在凝胶上形成不同的条带。但是此方法操作过于繁琐。
(5)传统杂交测序-芯片测序技术:传统杂交测序技术是基于印迹杂交或核酸原位杂交途径,利用寡核苷酸探针与目标dna杂交,其所携带的荧光基团会对dna进行标记,识别特异序列,其特征是测序读长比较短,该技术后来发展为芯片测序技术,是将具有特定碱基序列的探针固定在特殊的载体上,待测基因经提取、荧光标记后,与固定好的探针进行杂交,最后根据荧光的强度和种类测出待测序列的碱基类别。该类技术存在若干问题:只进行一次杂交,很难甄别非特异信号,因此可能由于一些非特异性杂交而导致序列误读,另外,芯片造价高昂,所需设备贵重,不利于普及应用。
(6)dhplc:目标核酸片段pcr扩增,部分加热变性后,含有突变碱基的dna序列由于错配碱基与正常碱基不能配对而形成异源双链。因包含错配碱基的杂合异源双链区比完全配对的同源配对区和固定相的亲和力弱,更易被从分离柱上洗脱下来,从而达到分离的目的。snps的有无最终表现为色谱峰的峰形或数目差异,依据此现象可很容易从色谱图中判断出突变的碱基。但是dhplc检测对所用试剂和环境要求较高,容易产生误差,不能检测出纯合突变。
(7)as-pcr技术:根据snp位点设计特异引物,其中一条链(特异链)的3′末端与snp位点的碱基互补(或相同),另一条链(普通链)按常规方法进行设计,因此,as-pcr技术是一种基于snp的pcr标记。因为特异引物在一种基因型中有扩增产物,在另一种基因型中没有扩增产物,用凝胶电泳就能够很容易地分辨出扩增产物的有无,从而确定基因型的snp。但是此方法也需要凝胶电泳。
(8)maldi-tofms:是将变性的单链pcr产物通过与硅芯片上的化合物共价结合后,在硅芯片上进行引物的退火,延伸反应,突变部位配对的碱基与正常配对的碱基不相同。根据引物在延伸反应中所结合的不同碱基的不同质量在质谱仪上显示不同峰而检测snp。
(9)实时荧光定量pcr技术,该技术由美国appliedbiosystems公司推出,所谓实时荧光定量pcr技术,是指在pcr反应体系中加入荧光基团,利用荧光信号积累实时监测整个pcr进程,最后通过标准曲线对未知模板进行定量分析的方法。目前荧光定量pcr应用领域广泛,一般用于目标模板定量或某一特定的核苷酸序列检测。在基因型别的检测方面,主要通过单通道的荧光信号值和ct(扩增循环数)值来判断是否为有效扩增。但是,pcr扩增步骤不仅对实验室设备要求过高,也为技术实施带来了极大的污染风险,从而严重影响检测结果的准确性。而且,当目标位点存在多个基因型时,特别是在单核苷酸多态性的检测时,单通道检测就不再实用,而需要使用双通道系统来检测目标位点的基因构成。对于双通道系统的结果,往往是分析两个通道的荧光曲线的ct值来判断是否为有效扩增从而确定为何基因型。这种方法对于纯合基因型和杂合基因型荧光曲线比较接近的时候容易出错,无法给出准确的结果,容易造成假阳性或误判。
鉴于现有技术在进行白细胞介素28b基因型的snp检测中存在的诸如误判率高、精准性差、稳定性低、检测效率低等技术问题,本发明意欲提供对现有技术进行改进,通过设计单链化衍生引导序列和探针序列,对各个组分的优化,提供一种精准性,稳定性,快捷性更高的snp荧光原位杂交测序检测方法,以为临床基因检测和个体化用药提供指导。
技术实现要素:
基于上述目的,本发明本发明公开了一种检测il-28b基因单核苷酸多态性的荧光原位杂交测序方法,所述方法具体包括如下步骤:
(1)提取待测样本的dna;
(2)以步骤(1)获取的dna为模板,在dntp、反应缓冲液、dna切割剂存在的情况下,在寡核苷酸分子的引导下进行单链化衍生,所述寡核苷酸分子分为第一引导序列和第二引导序列,同时加入两条序列各异的测序探针,分别被定义为第一测序探针和第二测序探针,在所述两条探针的5’端分别被标记了彼此不同荧光分子,在其3’端分别被标记了淬灭基团,所述第一引导序列、第二引导序列、第一测序探针和第二测序探针选自下列组合:
seqidno:1、2、7和8;或者
seqidno:3、4、9和10;或者
seqidno:5、6、11和12;
(3)对步骤(2)的单链化衍生物分别与第一测序探针和第二测序探针的杂交结果进行判读。
在一个优选的实施方案中,所述第一测序探针标记的荧光分子为5-羧基荧光素(fam),第二测序探针标记的荧光分子为六氯荧光素(hex),所述淬灭基团为bhq。
本发明提供的衍生引导寡核苷酸序列和探针序列的设计不同于一般的探针引物,探针对模板的识别性高。设计探针时荧光基团(fax和hex)在5端,猝灭基团在3端。由于snp位点的突变碱基只有一个,该技术在探针的设计上要针对性的对每一个snp位点进行独立的设计,并在后期的判读软件中进行精确分析,使整个发明准确、快速、经济的对dna序列和snp位点进行判读。
在一个优选的实施方案中,步骤(1)所述的待测样本为血液、体液、分泌物、代谢物、脱落物或组织样本。
在一个更为优选的实施方案中,所述待测样本dna模板浓度在103个/ul以上。
在一个优选的实施方案中,步骤(2)反应体系中第一引导序列与第二引导序列的比例为1:2-1:6,第二引导序列:第一测序探针:第二测序探针比例为1:1:1。
更为优选地,步骤(2)反应体系中第一引导序列与第二引导序列的比例为1:4。
所述单链化衍生体系中,所述dna切割剂可以为化学小分子切割剂、核酸内切酶、核酸外切酶、dna聚合酶等,在一个优选的实施方案中,所述切割剂为2.5u/μl的核酸外切酶,dntp为5mm,以及切割剂工作所需的10×缓冲液。
在另一个优选的实施方案中,步骤(3)的反应条件为:
(1)预变性95℃,10分钟,1个循环;
(2)变性95℃,30秒,60℃~64℃退火进行杂交,75秒,55个循环。
在又一个优选的实施方案中,步骤(3)所述判读的方法为读取衍生物分别与两种探针杂交后的荧光强度的st值(sequencingtimes,测序次数或测序深度,50次杂交即为50次杂交测序)之差。
其次,本发明还提供了一组用于检测il-28b基因型的核苷酸分子组合,所述组合包括单链化衍生中所用的定义为第一引导序列和第二引导序列的寡核苷酸分子、以及定义为第一测序探针和第二测序探针的杂交测序探针,所述第一引导序列、第二引导序列、第一测序探针和第二测序探针选自下列组合:
seqidno:1、2、7和8;或者
seqidno:3、4、9和10;或者
seqidno:5、6、11和12。
对于检测结果的判读方法,本发明通过解析荧光曲线,以杂交测序深度为基准分析荧光信号的变化规律,通过读取双通道荧光曲线的st值,从而直接判读出被测模板的dna序列和snp基因型。
与现有技术相比,本发明提供的杂交测序技术需要更多的模板核酸(通常不低于3000拷贝)才能完成测序,但是不需要经过pcr环节。这一特点具有独特的优势:(1)不易污染:少量异源dna污染的信号很低,不会影响测序准确性,例如100-300个拷贝的异源dna污染对pcr扩增而言是灾难性的,但是在杂交测序中,异源信号的强度不到总信号强度的10%,该信号可以作为本底信号而被去除;(2)简便:杂交测序不需要pcr扩增,因此在普通实验室即可开展,不需要pcr扩增实验室的强制认证;(3)简捷:杂交测序没有繁琐的标本dna提取和pcr扩增产物提取环节,将临床标本简单处理即可进行杂交测序,因此测序速度非常快,从拿到标本算起,2-3小时即可报告结果。
总之,本发明建立的检测il-28b基因单核苷酸多态性的荧光原位杂交测序方法,结合了杂交测序、荧光信号的采集、利用定义的st值进行分析运算并准确的给出被测样本的dna序列和基因型。本方法简洁易懂、使用方便、结果准确显示,非常有利于临床应用。
附图说明
图1a.荧光原位杂交测序检测单核苷酸多态性方法原理示意图;
图1b.荧光原位杂交测序检测snp结果判读示意图;
图1c.荧光原位杂交测序snp分析系统工作流程图;
图2.il-28b*1基因rs12979860的检测纯合cc基因型结果图;
图3.il-28b*1基因rs12979860的检测纯合tt基因型结果图;
图4.il-28b*1基因rs12979860的检测杂合ct基因型结果图;
图5.il-28b*2基因rs8099917的检测纯合gg基因型结果图;
图6.il-28b*2基因rs8099917的检测纯合tt基因型结果图;
图7.il-28b*2基因rs8099917的检测杂合tt基因型结果图;
图8.il-28b*3基因rs12980275的检测纯合gg基因型结果图;
图9.il-28b*3基因rs12980275的检测纯合aa基因型结果图;
图10.il-28b*3基因rs12980275的检测杂合ga基因型结果图。
具体实施方式
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但这些实施例仅是范例性的,并不对本发明的范围构成任何限制。
反应原理
与普通的taqman技术不同的是,本发明在杂交模板的制备上并没有采用pcr技术,而是采用了单链衍生引导寡核苷酸序列,使dna双链模板转化为单链衍生物,并直接与标记探针进行杂交测序。
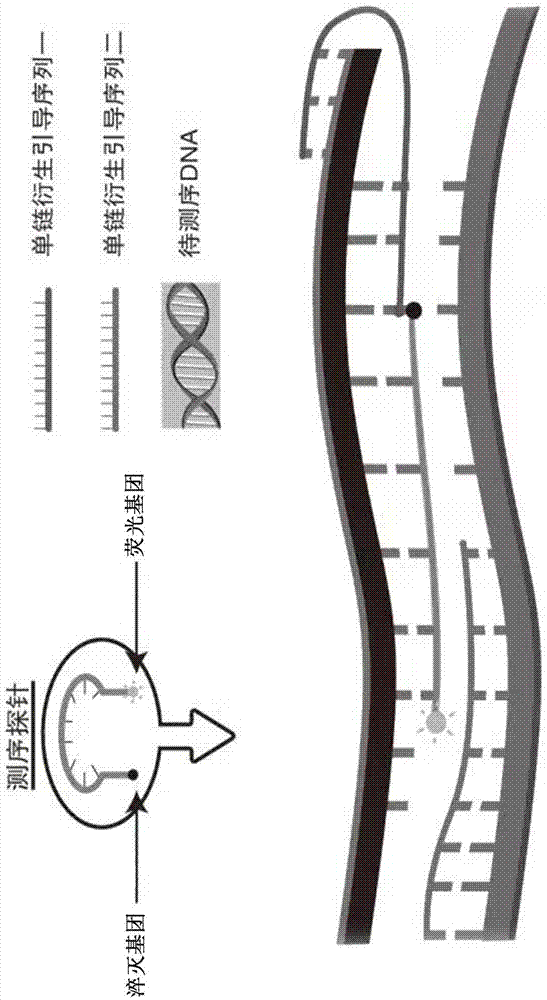
所述单链化衍生需要能够与模板链互补结合的寡核苷酸引导序列进行引发,引导序列可以在模板链的3’端和/或5’端与退火后的模板链特异性结合,并引发单链化衍生,所述引导序列和探针序列的设计原则是:针对特定的snp位点,设计2条具有在3’端和5’端特异性识别并结合目标snp的能力,并阻断特异性探针结合的模板链与其天然互补链的快速退火,进而维持模板单链衍生状态的寡核苷酸衍生引导序列、2条分别在5′端snp突变位点进行荧光修饰的探针。两种探针在其5′端前3个碱基以内设置snp突变核苷酸,分别使用不同荧光标记,并在探针另一端标记有淬灭基团。在探针与模板链结合前,探针保持因序列回折而形成的发夹结构,此时荧光基团因与淬灭基团在空间上毗邻而不能释放荧光。探针5′端上游4~15bp的模板序列为交叉区。单链衍生引导序列5′端具有交叉区互补的序列,此序列第一个碱基与探针的5′端第一个碱基不匹配,不匹配的碱基形成“碱基翘起”,导致探针的交叉区与单链衍生引导序列形成“发卡结构”,此结构的tm温度比探针高4~8℃,其能量一般为-8~-12kj。单链衍生引导序列与测序探针交叉区有6~12bp的间隔区域,用于保持“发夹结构”中的“环”区序列。“发卡结构”能够提高切割剂识别的准确性。当基于探针结合效率和dna聚合酶的核酸外切酶活性,实现与模板特异结合的探针的切割,从而释放相应的荧光信号。图1a为荧光原位杂交测序检测单核苷酸多态性方法原理示意图,由图中可以看出,在寡核苷酸探针的3端用荧光素标记(黑圈),在5端接有淬灭基团(黑点),在未杂交时,寡核苷酸探针是环状的,不发出荧光,当寡核苷酸探针与带有特异核苷酸snp位点(a)的模板结合时,探针的环状被打开,此时淬灭基团远离荧光素基团,荧光素基团发出荧光,荧光通过机器被检测出来。
在任何一次杂交测序中,两条测序探针与待测序的单链特异或非特异杂交,都会产生荧光信号。在累积一定次数的测序的荧光信号时,荧光信号强度可能达到某一数值。本发明的结果判读是通过计算释放出的荧光信号的st值,以及不同snp杂交测序之间的st值来进行的,所谓st值为sequencingtimes的缩写,即测序次数或测序深度,50次杂交即为50次杂交测序,每完成一次杂交测序,计算一次st值。此时,设定野生型测序探针a1的测序次数为sta1,设定突变性测序探针a2的测序次数为sta2。
st值之差:δst=sta1-sta2
在实际杂交测序检测中,需要用标准模板序列来设定每一次测序的阈值,按如下方式计算:
(1)投入野生型标准序列模板,此时获得的δst=sta1-sta2为:δst1
(2)同时投入野生型与突变性标准序列模板,此时获得的δst=sta1-sta2为:δst2
(3)投入野生型标准序列模板,此时获得的δst=sta1-sta2为:δst3
(4))st阈值的设定:
阈值1:取δst1与δst2之间的中位数(δst1+δst2)/2
阈值2:取δst2与δst3之间的中位数(δst1+δst2)/2
(5)判定:对于任何待测未知序列,计算其δst:
a、若在阈值1之外为野生纯和序列;
b、若在阈值2之外为突变纯和序列;
c、若在阈值1与2之间,为杂合序列。
在具体测定中,如图1b所示,图1b上图中,当野生型b1模板与野生型测序探针a1完全互补,与与突变型测序探针a2不完全互补。达到特定信号强度的测序深度st值,sta1小于sta2。这种情况下,b1模板为野生型基因型。
图1b中图中,当模板与测序探针a1和a2都完全互补,达到特定信号强度的st值,sta1与sta2很接近。这种情况下,模板为杂合序列。
图1b下图中,当b2模板与测序探针a2完全互补,与测序探针a12不完全互补。达到特定信号强度的测序深度st值,sta1大于sta2。这种情况下,b2模板为突变型基因型。
本发明还对实验体系组分的配置上进行了优化配置,通过不同浓度的衍生引导序列配制、优化配组的mg2+和dntps等,可以得到良好的检测结果。本方法高效快速,结果准确,能一次检测大量样本。由于没有扩增环节,极大程度避免了因样本污染而带来的污染放大效应进而影响了检测的假阳性,因此,具有极高的特异性。
检测实施例
首先针对snp位点序列,利用beacondesigner8设计单链衍生引导序列和探针序列。单链衍生引导序列和探针序列均由北京华夏时代基因科技发展有限公司合成。表1中,列出了检测用探针和单链衍生引导序列对应的snp检测位点。探针标记采用本领域常规标记技术即可。
表1:检测用探针和单链衍生引导序列对应的snp位点
从血液中进行白细胞的提取,将待检edta抗凝血液标本颠倒混匀数次;新抽取的edta抗凝血液标本,存于4℃,应在24小时内处理。红细胞裂解液(氯化铵)与灭菌水按1:9比例稀释为工作液。取1.5ml离心管,加入1ml红细胞裂解液(工作液),取150ul待检标本加入离心管中,上下颠倒混匀,室温静置5min;血样与红细胞裂解液混匀后,液体颜色变为澄清的红色。将离心管放入离心机中,离心5min;转速:3000转/700g。离心完成后取出离心管,用枪头吸去上清;离心后,离心管底部可见白色米粒大小状白细胞。加入100ulpharm-gene01snp分析保存液,混匀后,备用。
将第一/二单链衍生引导序列和第一/二测序探针干粉离心,3000rpm,1min,加超纯水溶解,稀释为20μm,利用紫外分光光度计sma1000测定260nm下的od值,
参照公式:
odseqidno:2/lseqidno:2×vseqidno:2=odseqidno:1/lseqidno:1×vseqidno:1×4,即引导序列一:引导序列二=1:4,
以及公式:
odseqidno:2/lseqidno:2×vseqidno:2=odseqidno:7/lseqidno:7×vseqidno:7=odseqidno:8/lseqidno:8×vseqidno:8,即引导序列二:第一测序探针:第二测序探针2=1:1:1,计算出反应体系中单链衍生序列和第一/二测序探针的体积。
反应体系
上述反应体系的配置也适用于其它序列组合。
在天隆tl988a荧光检测仪上进行反应,单链化衍生和杂交测序反应条件:过程1:预变性95℃,10min,1个循环过程2:变性95℃,30s,62℃退火进行杂交,75s,共进行55个杂交测序过程循环。
在荧光检测仪(天隆tl988a)上进行检测,在整个单链化衍生和杂交测序过程中,第一探针和第二探针分别与模板结合,匹配的特异结合后发出荧光信号,不匹配的非特异结合可能会发出很微弱的信号或者不发出信号,根据荧光强度来做出判断。为了便于机器自动判读,将图1b中的原理,转化为阈值阐述,从而可设定程序,由软件自动判读序列。
本发明对检测结果的判读分析是通过“荧光原位杂交测序snp分析系统”进行的,所述系统包括工作控制模块、后台设置模块、样本设置模块、温控设置模块、运行监控模块和数据分析模块。所述“荧光原位杂交测序snp分析系统”是北京华夏时代生物工程有限公司设计编写的。
工作控制模块主要包括新建程序、打开程序、保存结果、开始或结束程序以及打开后台设置界面等功能。
后台设置模块主要为参数设置,主要参数包括基因类型、通道1st值上限、通道1st值下限、通道2st值上限、通道2st值下限、st值差上限、st值差下限、通道1基因型、通道2基因型和温控条件选择。
样本设置模块主要包括样本名称、基因类型和结果。
温控设置模块则是不同温控条件设置界面,根据实际情况设定不同的温控程序。
运行监控模块主要包括温控监控板块和荧光读取板块,实时显示随着杂交测序的进行,其荧光值的变化曲线。数据分析界面主要显示耦合后的荧光检测曲线,同时读取并显示双通道荧光曲线的st值,进行运算后显示该样本的基因序列和基因型结果。
数据分析模块显示耦合后的荧光检测曲线,同时读取并显示双通道荧光曲线的st值,进行st值差的运算后显示该样本的基因型结果。st值差是通道一st值减通道二st值得到的数值,在软件后台会根据对标准序列杂交测序结果,设置一个标准st值,将检测样本杂交测序所得的st值与标准st值比对,即可判断待测样本的基因序列。
图1c显示了本发明“荧光原位杂交测序snp分析系统”的运行示意图。
程序运行完成后,在数据分析模块界面,会显示双通道(标记fam的野生型探针通道和标记hex的突变型探针通道)荧光曲线st值,根据后台设置模块设置的参数,软件会自动计算并给出检测样本的基因序列和基因型。在样本设置界面,选择需要打印检测结果的样本名,然后点击打印即可得到最终的检测结果。
按照两个通道的荧光数据采集的大小,根据st值得差值,系统自动给出snp位点的基因序列和基因型。软件分析判读结果的方法有两种:1.根据荧光强度来判断:系统检测两条荧光通道,通道一(fam通道)和通道二(hex通道),系统首先计算检测荧光的荧光强度。如果计算出来的荧光强度大于软件里设置的噪声荧光的数值,系统认为这个通道的荧光数据是有效的,进而计算st值;如果计算的荧光强度小于设置的噪声荧光的数值则认为是无效的数据,略过,st会显示成0或者-1。如果两个通道的荧光强度均小于所设置的数值,软件显示为“重测”。2.根据st差值来判断:软件默认是通道一(fam通道)st值减去通道而(hex通道)的st值。当得出的st差值在软件设置的杂合基因序列st阈值上下限之间时,结果显示为杂合。当st差值在设置的st值上下限之外时,结果显示为st值小的那个通道所代表的基因型。整个程序运行完毕需要2个小时31分钟。
图2-图10为具体snp检测的判断结果。
图2为il-28b*1基因rs12979860的检测纯合cc基因型结果图,在图中有两条直线代表着两条通道,各携带荧光标记的探针,通道一(黑色线标记)探针标记cc基因型,通过前述原理,与标准模板的st阈值比对,可计算出此样本的基因型为cc基因型。
图3为il-28b*1基因rs12979860的检测纯合tt基因型结果图,在图中有两条直线代表着两条通道,各携带荧光标记的探针,通道二(浅灰色线标记)探针标记tt基因型,通过前述原理,与标准模板的st阈值比对,可计算出此样本的基因型为tt基因型。
图4为il-28b*1基因rs12979860的检测杂合ct基因型结果图,在图中有两条直线代表着两条通道,各携带荧光标记的探针,通道一(黑色线标记),通道二(浅灰线标记)探针标记cc和tt基因型,通过前述原理,与标准模板的st阈值比对,可计算出此样本的基因型为ct基因型。
图5为il-28b*2基因rs8099917的检测纯合gg基因型结果图,在图中有两条直线代表着两条通道,各携带荧光标记的探针,通道一(黑色线标记)探针标记gg基因型,通过前述原理,与标准模板的st阈值比对,可计算出此样本的基因型为gg基因型。
图6为il-28b*2基因rs8099917的检测纯合tt基因型结果图,在图中有两条直线代表着两条通道,各携带荧光标记的探针,通道二(浅灰色线标记)探针标记tt基因型,通过前述原理,与标准模板的st阈值比对,可计算出此样本的基因型为tt基因型。
图7为il-28b*2基因rs8099917的检测杂合gt基因型结果图,在图中有两条直线代表着两条通道,各携带荧光标记的探针,通道一(黑色线标记),通道二(浅灰线标记)探针标记gg和tt基因型,通过前述原理,与标准模板的st阈值比对,可计算出此样本的基因型为gt基因型。
图8为il-28b*3基因rs12980275的检测纯合gg基因型结果图,在图中有两条直线代表着两条通道,各携带荧光标记的探针,通道一(黑色线标记)探针标记gg基因型,通过前述原理,与标准模板的st阈值比对,可计算出此样本的基因型为gg基因型。
图9为il-28b*3基因rs12980275的检测纯合aa基因型结果图,在图中有两条直线代表着两条通道,各携带荧光标记的探针,通道二(浅灰线标记)探针标记aa基因型,通过前述原理,与标准模板的st阈值比对,可计算出此样本的基因型为aa基因型。
图10为il-28b*3基因rs12980275的检测杂合ga基因型结果图,在图中有两条直线代表着两条通道,各携带荧光标记的探针,通道一(黑色线标记),通道二(浅灰线标记)探针标记gg和aa基因型,通过前述原理,与标准模板的st阈值比对,可计算出此样本的基因型为ga基因型。
序列表
<110>北京华夏时代生物工程有限公司
<120>检测il-28b基因单核苷酸多态性的荧光原位杂交测序方法
<160>12
<170>patentinversion3.3
<210>1
<211>29
<212>dna
<213>homosapiens
<400>1
tcgcsttcgggcstgtcgtgtastgaacc29
<210>2
<211>19
<212>dna
<213>homosapiens
<400>2
ggtcaatcacagaagggag19
<210>3
<211>31
<212>dna
<213>homosapiens
<400>3
atgstcacagatgtcastgttcstcsttttg31
<210>4
<211>20
<212>dna
<213>homosapiens
<400>4
tgggagaatgcaaatgagag20
<210>5
<211>29
<212>dna
<213>homosapiens
<400>5
attstaggaatcstacatgaggtgstgag29
<210>6
<211>22
<212>dna
<213>homosapiens
<400>6
tstgtacccattaaacaacaac22
<210>7
<211>16
<212>dna
<213>homosapiens
<400>7
cgaaccagggttgaat16
<210>8
<211>18
<212>dna
<213>homosapiens
<400>8
tgaaccagggttgaattg18
<210>9
<211>20
<212>dna
<213>homosapiens
<400>9
atttcacccaaattggaacc20
<210>10
<211>19
<212>dna
<213>homosapiens
<400>10
atgtcacccaaattggaac19
<210>11
<211>22
<212>dna
<213>homosapiens
<400>11
acagacgtgtstaaatatttgc22
<210>12
<211>20
<212>dna
<213>homosapiens
<400>12
acggacgtgtstaaatattt20
- 还没有人留言评论。精彩留言会获得点赞!
- CYP2C9*3基因多态性快速检测的引物、分子信标、试剂盒及其检测方法与流程
- 一种单核苷酸多态性位点在辅助鉴定大豆花叶病毒抗性中的应用的制造方法与工艺
- DHFR基因多态性检测体系及其试剂盒的制造方法与工艺
- ERCC1基因多态性检测体系及其试剂盒的制造方法与工艺
- 一种判定中国人群rs125055单核苷酸多态性与儿童IBD之间的相关性的方法与制造工艺
- 黄牛ADNCR基因单核苷酸多态性的四引物扩增受阻突变体系PCR检测方法及其应用与制造工艺
- 使用PNA探针的熔解曲线分析、使用熔解曲线分析用于分析核苷酸多态性的方法和试剂盒与制造工艺
- 人KRAS中单核苷酸多态性的检测的制造方法与工艺
- 一种InDel位点基因型分型的方法与制造工艺
- 单核苷酸多态性rs145562243在筛查麻风患者中的应用的制造方法与工艺
- CYP2C9*3基因多态性快速检测的引物、分子信标、试剂盒及其检测方法与流程
- 一种单核苷酸多态性位点在辅助鉴定大豆花叶病毒抗性中的应用的制造方法与工艺
- DHFR基因多态性检测体系及其试剂盒的制造方法与工艺
- ERCC1基因多态性检测体系及其试剂盒的制造方法与工艺
- 一种判定中国人群rs125055单核苷酸多态性与儿童IBD之间的相关性的方法与制造工艺
- 黄牛ADNCR基因单核苷酸多态性的四引物扩增受阻突变体系PCR检测方法及其应用与制造工艺
- 使用PNA探针的熔解曲线分析、使用熔解曲线分析用于分析核苷酸多态性的方法和试剂盒与制造工艺
- 人KRAS中单核苷酸多态性的检测的制造方法与工艺
- 一种InDel位点基因型分型的方法与制造工艺
- 单核苷酸多态性rs145562243在筛查麻风患者中的应用的制造方法与工艺
- 一种用于评估皮肤防御情况的引物和探针及试剂盒的制作方法
- 一种用于评估皮肤防晒情况的引物和探针及试剂盒的制作方法
- 一种用于评估皮肤抗皱情况的引物和探针及试剂盒的制作方法
- 人PLIN1基因rs2289487位点多态性检测方法及试剂盒的制作方法
- 测定人NEFM基因rs12515位点多态性的方法及试剂盒的制作方法
- 一种人TERF1基因rs3863242位点多态性检测技术的制作方法
- 测定人CCNH基因rs3093816位点多态性的方法及试剂盒的制作方法
- 测定人NEFL基因rs2979704位点多态性的方法及试剂盒的制作方法
- 测定人PON1基因rs854560位点多态性的方法及试剂盒的制作方法
- 测定人PON1基因rs662位点多态性的方法及试剂盒的制作方法