DNA甲基转移酶及其可溶性异源表达和分离纯化方法与流程
本发明属于分子生物学技术领域,涉及一种dna甲基转移酶及其可溶性异源表达和分离纯化方法。
背景技术:
dna甲基化是一种广泛存在且非常重要的dna表观遗传机制,对基因的表达、基因组的稳定性以及细胞的分化等具有重要的调节作用。介导催化dna甲基化修饰的酶主要是dna甲基化转移酶,广泛分布于所有原核和真核生物生命体中,可特异性识别和修饰基因组特定序列中的特定碱基位置。在几乎所有细菌或原核生物中,dna甲基转移酶常与识别同一位点的限制性内切酶组成细菌的初级免疫防御系统—限制修饰系统,保护宿主细胞免受外来基因组的侵袭,从而维持自身生命活动或遗传稳定性。同时,还参与了调控dna的复制、修复以及共生与致病性等生理活动。在真核系统中,dna甲基转移酶具有非常重要的表观遗传调控作用,可参与调控基因转录、细胞分化、胚胎发生、基因组印记以及可移动遗传元件活动等。因此,dna甲基转移酶正常功能的紊乱,会对哺乳动物胚胎发生的产生致命结果以及对植物生长发育的产生重大障碍。
作为分子和细胞生物学研究的强有力工具,dna甲基转移酶已广泛应用于基因工程改造、分子生物学实验甚至是细菌感染的药物靶点等重要领域。将外源dna甲基转移酶转化真核细胞,实现dna甲基转移酶的异源表达,使其对真核基因组进行甲基化已成为体内分析功能性染色质结构以及研究dna甲基化在细胞过程中功能作用的最有前景的方法。同时,利用dna甲基转移酶融合dna结合蛋白技术开发特异性遗传基因座靶向甲基化研究的新方法,也将有益于生命科学领域的研究。因此,开发利用dna甲基转移酶,研究其空间结构及分子作用机制,不仅可为表观遗传治疗提供前景,而且可为解决重大科学问题提供强有力的工具和思路。
然而,dna甲基转移酶的异源表达及分离纯化面临着诸多挑战,首当其冲的是其本身的蛋白毒性。细胞为维持自身基因组dna的稳定性,保护其免受内源性限制性内切酶切割,外源dna甲基转移酶异源诱导表达时会大量形成包涵体,不利于其分离纯化及后续研究。因此,选择合适的重组表达载体及宿主、优化诱导条件和分离纯化方法就显得尤为重要。本发明所涉及的dna甲基转移酶来源于耐辐射奇球菌,是一种n4-cytosinedna甲基转移酶,其异源克隆表达及分离纯化方法尚未见报道。由于耐辐射奇球菌是一种对辐射和干旱等极端条件具极强抗性的微生物,其体内的很多蛋白活性需要独特的生理条件,因此该细菌中dna甲基转移酶的表达和纯化过程显得尤为困难,需要付出特别艰辛的劳动和智慧。我们尝试了大量已报道的dna甲基转移酶表达和纯化方法,均不能进行体外表达,或以包涵体的形式存在,无法获得有活性的纯化蛋白。
技术实现要素:
为了克服现有技术的不足,本发明的目的是提供一种dna甲基转移酶及其可溶性异源表达和分离纯化方法。
一种dna甲基转移酶,编码所述dna甲基转移酶的核苷酸序列包括:
1)如seqidno.1所示的核苷酸序列;
2)与seqidno.1所示的核苷酸序列不同,但编码相同氨基酸序列的核苷酸序列;
3)与seqidno.1所示的核苷酸序列具有80%以上的同源性,且编码的蛋白具有n4-cytosinedna甲基转移酶活性的核苷酸序列;
4)与1)、2)和3)中的核苷酸序列互补或杂交的核苷酸序列;
所述的dna甲基转移酶属α型n4-cytosinedna甲基转移酶,可识别dna保守序列为5’-ccgcgg-3’,并特异性甲基化修饰第二个胞嘧啶c的n4位,产生4mc类型甲基化修饰。
一种重组表达载体,含有所述的编码dna甲基转移酶的核苷酸序列的重组原核表达载体。
一种原核表达宿主细胞,所述宿主细胞转化了所述的重组表达载体或者宿主细胞基因组中整合了所述的核苷酸序列的宿主细胞及其繁殖后代细胞。
一种dna甲基转移酶的可溶性异源表达方法,所述dna甲基转移酶的可溶性原核表达步骤为:将所述的原核表达宿主细胞活化2次,扩大培养至o.d.6000.6,冰水浴冷却,无菌加入0.4-0.6mmiptg,16℃,220rpm诱导培养20h。
所述的dna甲基转移酶的可溶性异源表达方法,所述的诱导培养后收集细胞进行破碎,步骤如下:先在4℃、800-1200bar条件下高压破碎2-4min;再在冰水浴中,用功率60%、超声3s和间隙9.9s超声破碎60-90min;最后低温高速离心30-50min,收集上清。
一种所述的dna甲基转移酶的分离纯化方法,收集异源表达后的细胞破碎得到上清后进行dna甲基转移酶的分离纯化,步骤依次为:ni柱层析,tev蛋白酶酶切,mbp柱,脱盐,ni柱层析,免疫印迹鉴定,再脱盐,heparin柱,分子筛。
所述的分离纯化方法,进一步进行质谱鉴定,酶活性分析。
本发明的有益效果:
本发明提供了一种未被报道过的dna甲基转移酶,克隆表达及其分离纯化方法,可为其它限制性内切酶或者dna甲基化酶的开发应用提供方法学参考,对开发新型分子生物学的工具酶具有重要意义。
附图说明
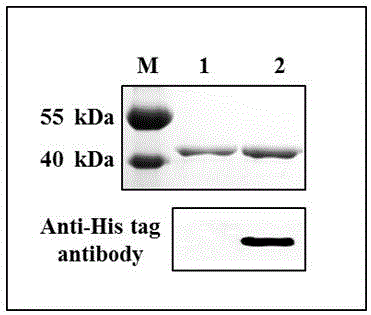
图1是m.drar1甲基转移酶western-blot检测结果图。泳道1为m.drar1目的蛋白,经tev蛋白酶酶切后不含6×his标签,大小约为48.7kda,wb检测结果为阴性;泳道2为tev蛋白酶酶切后所分离的his-mbp蛋白,大小约为44.7kda,含6×his标签,wb结果呈阳性。
图2是m.drar1甲基转移酶最后经分子筛纯化及sds-page电泳的结果图。所用分子筛型号为superdex7510/300g,m.drar1目的蛋白从进样到出峰的洗脱体积约为10ml,表明其在溶液中是以单体形式存在,sds-page电泳结果显示其纯度在98%以上。
图3是m.drar1甲基转移酶质谱检测结果。质谱结果显示所测目的蛋白绝大多数氨基酸序列均与参考序列值相同,其中加粗黑体字体显示质谱检测出的肽段,灰色代表尚未检出的肽段。
图4是m.drar1甲基转移酶的酶活分析。其中,a为m.drar1体外甲基化修饰λdna酶切结果图;b为m.drar1体外甲基化修饰λdnapcr扩增片段酶切结果图;泳道1和4为原始底物,泳道2和5为未被甲基化阴性对照,泳道3和6为甲基化修饰阳性结果。
具体实施方式
以下结合附图和具体实施例对本发明做进一步的阐述。
实施例1:dna甲基转移酶m.drar1的克隆及表达载体的构建
为获得可溶性目的蛋白,本发明优化筛选了多种表达载体(包括pet-28a、pet-22b、prrs和pet28a-hmt等)、宿主(包括bl21、bl21(de3)、bl21(de3)plyss、transetta(de3)、transb(de3)及e.colier2566等)及诱导表达条件(如iptg浓度、诱导温度和时间等),最终选择可溶性表达良好的pet28a-hmt与e.colier2566宿主作为原核表达体系(见表1)。
(1)使用天根生物的细菌基因组dna提取试剂盒(dp302-02)提取耐辐射奇球菌基因组dna,nanodrop1000(美国thermo公司)测定dna浓度和纯度。针对dna甲基转移酶m.drar1的编码基因序列设计一对全长特异性引物,同时在每条引物的5’端引入与载体相同的限制性酶切位点,具体引物序列如下:
(2)上游引物m.drar1-f:5’-ttaatttcatatgacgcaacctcttctctttgacc-3’(seqidno.2),其中,单下划线为ndei限制性酶切位点,双下划线为对应的保护碱基。
(3)下游引物m.drar1-r:5’-tatggatccttacctggtcagttcaaccacgg-3’(seqidno.3),其中,单下划线为bamhi限制性酶切位点,双下划线为相应的保护碱基。
(4)以耐辐射奇球菌基因组dna为模板,采用北京全式基因公司的transstart®fastpfudnapolymerase(ap221)体外pcr扩增,pcr扩增体系及条件按照该试剂说明书设置,扩增结束经电泳检测合格后,采用lifescience公司的agarosegelextractionkit/pcrcleankit(catno.116401)纯化回收pcr扩增产物。
(5)表达载体的选择:为增加目的蛋白的可溶性表达,本发明从pet-28a、pet-22b、pet-20b、prrs(neb公司roberts教授馈赠)和pet28a-hmt(即在pet28a质粒核糖体结合位点rbs后插入一段融合n端6×his标签、麦芽糖结合蛋白mbp标签以及tev蛋白酶酶切位点的碱基序列,本实验室保存)等多种质粒通过后续可溶性表达测试的方法筛选表达载体,最终选择pet28a-hmt为目的蛋白可溶性表达载体(见表1)。
表1是本发明优化筛选所用的原核表达载体、宿主、诱导条件及诱导结果和可溶性分析统计。其中“/”表示因蛋白表达量过低无法进行诱导测试及可溶性分析。
表1
(6)将(4)中pcr扩增产物和(5)中pet28a-hmt质粒均用限制性内切酶ndei和bamhi(takara公司)双酶切过夜,酶切体系和温度参考试剂说明书。酶切结束后电泳检测,胶纯化回收双酶切产物。使用t4dna连接酶(takara公司)将酶切后的目的片段与质粒在16℃条件下连接过夜,按转化操作说明将链接产物直接转化e.colidh5α感受态细胞(全式基因,cd201),吸取50-100μl复苏后的细胞均匀涂布于lk固体培养基皿(lb固体培养基含40-50μg/ml卡那霉素),37℃倒置培养过夜,挑选若干单克隆子于5mllk液体培养基中,37℃,220rpm震荡培养20h左右。
(7)采用美国axygen公司质粒提取试剂盒(cat.no.ap-mn-p-250g)按操作说明提取质粒,nanodrop1000测定质粒纯度和浓度,首先使用ndei和bamhi限制性内切酶进行双酶切鉴定,将鉴定成功的重组质粒再进一步测序验证,所用上游测序引物hmt-f如seqidno.4所示,下游测序引物hmt-r如seqidno.5所示,测序结果进行blast在线比对,测序正确的质粒即为构建成功的重组表达质粒。
实施例2:dna甲基转移酶m.drar1的原核表达
(1)大肠杆菌e.colier2566感受态细胞的制备:将e.colier2566菌株(zonhonbiopharmainstitute,inc.,zhr5015)在无任何抗性lb固体培养皿上划线活化后,挑取单克隆接种于5mllb液体培养基,37℃,220rpm震荡过夜培养。按照《分子克隆指南》中感受态细胞制备方法无菌操作制备er2566感受态细胞,100μl分装后置于-80℃超低温冰箱中保存备用。
(2)pet28a-hmt-m.drar1重组载体的转化:从-80℃超低温冰箱中取er2566感受态细胞于冰上解冻,无菌加入10-20μg重组表达载体,轻混,冰浴30min,42℃水浴热激45-90s,然后再冰浴2-3min,加入500μl无菌lb液体培养基后置于37℃,220rpm震荡培养约1h,使菌株充分复苏。取50-100μl复苏细胞均匀涂布于lk固体培养基皿,37℃倒置培养过夜,取若干单克隆子菌落pcr验证重组质粒是否成功进入表达宿主,所用引物对如实施例1(2)和实施例1(3)所示。
(3)m.drar1蛋白的诱导表达:将(2)中的表达菌株接种至5mllk液体培养基中,37℃,220rpm震荡培养过夜。取1ml培养好的菌株再接种至100mllk液体培养基中,37℃,220rpm震荡培养至o.d.600=0.6~0.8,立刻置于冰水浴冷却,5ml分装至50ml无菌离心管中,分别加入不同浓度的iptg(0、0.2、0.4、0.6和1.0mm),再分别置于不同温度,220rpm震荡诱导培养,其中,16℃诱导培养20h,20℃诱导16h,30℃诱导5h,37℃诱导3h。诱导结束后,按0.3/o.d.600ml的比值取样,12000rpm离心1min收集菌体,加入30-50μl1×sdsloadingbuffer重悬细胞,沸水浴煮沸10-15min至蛋白完全变性,15000rpm离心3-5min,各吸取8-10μl上清样本,sds-page凝胶电泳观测诱导结果,确定最佳的iptg诱导浓度为0.4mm,诱导温度范围为16-37℃。
(4)m.drar1甲基转移酶的可溶性表达:按(3)的方法诱导培养表达菌株,按0.4-0.6/o.d.600ml的比值取样,12000rpm离心收集菌体。加入60μl裂解缓冲液(20mmtris-hclph8.0,500mmnacl,3mmβ-me)重悬细胞。悬浮细胞先用液氮迅速冷冻30s,然后再37℃水浴溶解5min,如此反复冻融5次以上,溶液呈澄清透明状可确保细胞完全溶解。15000rpm高速离心20-30min后将上清和沉淀完全分离,并做好标记。将上清液中加入15μl5×sdsloadingbuffer,沉淀中加入75μl1×sdsloadingbuffer,混匀后沸水浴煮沸5-10min至蛋白完全变性,sds-page凝胶电泳检测目的蛋白在不同诱导温度下的可溶性,进一步确定最佳诱导条件为温度16℃诱导培养20h。
(5)m.drar1蛋白的可溶性测试:按(3)和(4)的方法诱导培养目的蛋白和取样。分别60μl加入不同的裂解buffer(20mmtris-hclph7.0-8.0,50-1000mmnacl,3mmβ-me)重悬细胞。按(4)的方法制备可溶性测试样本,sds-page电泳检测目的蛋白在不同盐浓度和ph条件下的可溶性,结果表明m.drar1蛋白的最适可溶性buffer为20mmtris-hclph7.8-8.0、50-500mmnacl和3mmβ-me。
实施例3:dna甲基转移酶m.drar1的分离纯化
(1)按实施例2(3)和实施例2(4)方法诱导培养目的蛋白,诱导结束后,8000rpm,4℃,10min离心收集菌体,1×pbs溶液洗涤一次,再次离心收集菌体,-80℃保存备用。按20:1的比例,即1g菌体(湿重)加入20ml裂解buffer(20mmtris-hclph8.0,500mmnacl,3mmβ-me,5%glycerol,9mmimidazole)悬浮细胞,冰浴冷却5-10min。
(2)细胞破碎:以上悬浮细胞先采用高压均质机(上海励途,fb-110系列)高压破碎细胞,参数为4℃、800-1200bar、时间2-4min。再继续用冰水浴超声破碎细胞(宁波新芝,jy92-iin),参数为功率60%、超声3s、间隙9.9s、时间60-90min。超声结束后,4℃,15000rpm,30min高速离心,收集上清,冰浴备用。
(3)镍柱亲和纯化:将制备的上清经0.22或0.45μm微孔滤膜过滤,再利用akta蛋白纯化系统(美国ge公司)分离纯化目的蛋白。先用镍柱(1mlni-nta,ge公司)亲和层析纯化,其中,ni-buffera为20mmtris-hclph8.0、500mmnacl、3mmβ-me和5%glycerol,ni-bufferb为20mmtris-hclph8.0、500mmnacl、3mmβ-me、5%glycerol和500mmimidazole。纯化参数为进样流速1ml/min、洗脱流速2ml/min、7%bufferb去除杂蛋白以及50%bufferb洗脱收集目的蛋白,nanodrop测定蛋白浓度。
(4)tev蛋白酶酶切去除标签:tev蛋白酶由本实验室纯化并保存,将其以上收集的目的蛋白按照1:100的质量比例混合,反应体系为20mmtris-hclph8.0、100-500mmnacl、5%~10%glycerol、0.5mmedta和1mmdtt,4℃酶切过夜。
(5)mbp柱纯化:采用akta蛋白纯化系统,将上述酶切后的样本进一步纯化,5ml柱载量的mbp纯化柱购自ge公司,纯化所用mbp-buffera为20mmtris-hclph8.0、500mmnacl、5%glycerol和1mmdtt,mbp-bufferb为20mmtris-hclph8.0、500mmnacl、20mmmaltose、5%glycerol和1mmdtt,纯化参数为进样流速0.5ml/min、收集全部流穿液。100%mbp-bufferb和2ml/min流速洗脱mbp标签及含mbp标签的目的蛋白,收集备用。
(6)脱盐柱脱盐:使用15ml超滤管(millipore公司,30kda)将以上收集的流穿液浓缩至10ml左右体积,再经脱盐柱脱盐处理,脱盐buffer为20mmtris-hclph8.0、200mmnacl和5%glycerol,脱盐参数为进样流速1ml/min、洗脱流速2ml/min、收集目的蛋白,约24ml。
(7)镍柱进一步亲和纯化:将获得的目的蛋白溶液按(3)中所述方法进一步纯化,目的是进一步去除杂蛋白(例如his-tev蛋白酶和his-mbp蛋白等)。纯化参数为进样流速1ml/min、洗脱流速2ml/min、收集全部流穿液及3-5%bufferb洗脱液。
(8)western-blot检测:所得目的蛋白样本(不含his标签)和(5)中收集的杂蛋白样本(含his标签)经sds-page电泳后,免疫印迹法(western-blot)检测蛋白中his标签的含量,采用6×hisanti-histag小鼠单克隆抗体(proreintech公司,66005-1-lg)一抗,hrp标记山羊抗小鼠二抗(wuhanservicebiotechnology公司,gb23301),x光片自显影技术检测,结果如图1所示,表明所获目的蛋白不带任何his标签,为后续纯化和质谱分析所用。
(9)按(6)中所述方法进一步脱盐,脱盐buffer为20mmtris-hclph8.0、100mmnacl和5%glycerol。
(10)heparin柱纯化:所用heparin柱购自美国ge公司,纯化所用buffer-ha为20mmtris-hclph8.0、100mmnacl和5%glycerol,buffer-hb为20mmtris-hclph8.0、1000mmnacl和5%glycerol。纯化参数为进样流速1ml/min、洗脱流速2ml/min、15%buffer-hb洗脱杂蛋白、50%buffer-hb洗脱收集目的蛋白。
(11)分子筛superdex75纯化:所获目的蛋白用超滤管浓缩到约500μl体积后,采用分子筛柱进一步纯化目的蛋白,分子筛buffer为20mmtris-hclph8.0、200mmkcl,纯化参数为0.75ml/min进样和洗脱,计算进样到目的蛋白出峰的洗脱体积(约9.5ml),收集目的蛋白后经sds-page电泳检测蛋白纯度,结果显示目的蛋白纯度达98%以上,且以单体形式存在(如图2所示)。采用nanodrop仪器测定蛋白浓度,目的蛋白浓缩到一定浓度后分装,液氮速冻后于-80℃保存备用。
(12)目的蛋白质谱分析:将上述目的蛋白小心从sds-page胶中切割下来,切成小块后加少许ddh2o于低温保存后用maldi-tof进行质谱分析,结果表明蛋白大小及氨基酸序列均与理论值相同(如图3所示)。
实施例4:m.drar1蛋白的酶活性分析
(1)λdna片段的pcr扩增:采用pfu高保真聚合酶对λdna片段进行体外扩增,上游引物λdna-f序列如seqidno.6所示,下游引物λdna-r序列如seqidno.7所示,该片段含3个“ccgcgg”保守序列,全长3008bp。扩增结束后,经电泳检测合格后纯化备用(见图4b)。
(2)dna甲基转移酶m.drar1体外反应体系:将λ噬菌体dna2-5μg和上述pcr反应片段1-2μg加入含50-200mmkcl、10-50mmtris-hcl(ph7.5-8.0)、0.1mmedta、3-7mmβ-me及20-100μmsam的反应buffer中,另外加入1μm实施例3(11)的目的蛋白,置于30℃条件下反应1h,纯化备用(图4)。
(3)限制性内切酶sacii酶切反应:将纯化底物dna用限制性内切酶sacii(takara公司)按试剂使用说明进行切割,反应结束后经0.7%和1.0%琼脂糖电泳检测,结果进一步表明m.drar1甲基转移酶可甲基化修饰“ccgcgg”保守基序,保护底物免受限制性内切酶sacii的切割。
本发明实施例中所用的方法是一种分离纯化dna甲基转移酶的特有方法,根据本发明的教导和启示,任何使用相同或者类似方法纯化其它dna甲基转移酶,也在本发明的保护范围之内。
序列表
<110>浙江大学
<120>dna甲基转移酶及其可溶性异源表达和分离纯化方法
<141>2018-10-31
<160>7
<170>siposequencelisting1.0
<210>1
<211>1305
<212>dna
<213>耐辐射奇球菌(deinococcusradiodurans)
<400>1
atgacgcaacctcttctctttgacctgccaacgccacgacctacctaccgggatactgcc60
ttcgcttccaacaagacgctggcgatgcaccgttgggtgaactggattgcgggattctcc120
tctgagtttgtgcagcacgctcttgagctgcatcttcctgaccccaaccctgagcaagtc180
gttctagaccccttcggcggcgttggtaccacaccgattaccgcctttctgcgaggccac240
tctgtcgtctcctacgacatcaacccctttcctctgttggtgcagcgggccaagctcaga300
gctattcaggacgtgactcctgctgaatttgcacagcagattgaagcgttcacagctcac360
atggcgactggaggagtacccaagagcaaggtgccccagggctttacctcgcggacgccc420
ttctacagcgagaaggtgctggtgaaggtgctgcacgtttgggacttcatcaacgaggtg480
gccgatgaagacctgcgcgacctgttccaggtggccttcggagccacgatggtgtcctac540
tcgaactattcctatgagccgtcattgggttccagggcggctgccgggaaacctgacatc600
gaggacgctgacgtggcccaggtcatgcgggacaagctgctggagatgcacgccgaccta660
ttaggcgtgcagggcatcaagttgggtggccagaccgctcaggtgtaccaagggtctttc720
atgcgttcagagctacccgactccagcgttgacttgatggtgacctcgccgccctacctg780
aacaactatcactacctgcggaatacccggcctcacctttactggctgggctacgctacc840
agcccgaaggacttgcggtacttggaattggacaactacggcaaatactggcagaccgtg900
cgagacgcgaagtatcagacctcactgatatttgattcaccttggctgcaagacctggtg960
aaccagctcgcgggcgttcagtcagatagaggcgtgtatggcggccaaggatgggcgaac1020
tatgccactgagtatttcaacgacacctaccgcttcttgcagaagacgcaagctgtattg1080
cgtcctggcgcgaaggccctgattgtcgtcggcaactctatcgtcaaaggtacaaacctg1140
cctattgacgaggtattcacccacatcgctcagcacttgggcttcagcggccacgacatc1200
cacatggtgcgtgattcccgcatcggctcgagcattgtggggactgggctacggtctgag1260
gggaaagggagactgtacgaggccgtggttgaactgaccaggtaa1305
<210>2
<211>35
<212>dna
<213>人工序列(artificialsequence)
<400>2
ttaatttcatatgacgcaacctcttctctttgacc35
<210>3
<211>32
<212>dna
<213>人工序列(artificialsequence)
<400>3
tatggatccttacctggtcagttcaaccacgg32
<210>4
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>4
tgccgccactatggaaaacg20
<210>5
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>5
tcctttcgggctttgttagcag22
<210>6
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>6
tatgagagcctgcgtggacg20
<210>7
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>7
ttccggtaacggaccgagt19
- 还没有人留言评论。精彩留言会获得点赞!