无细胞DNA染色质免疫沉淀的诊断应用的制作方法
相关申请的交叉引用
本申请要求2018年3月13日提交的美国临时专利申请号62/642,158和2018年5月6日提交的美国临时专利申请号62/667,528的优先权权益,其整体通过引用并入本文。
本发明属于无细胞dna-蛋白质复合物分析领域。
背景技术:
细胞死亡(通过凋亡或坏死)后,短dna片段被释放到血浆中。这些通常被称为循环无细胞dna(cfdna)或循环肿瘤dna(ctdna,如果源自肿瘤细胞)。cfdna的存在已被认知数十年,其中cfdna片段的一般长度为约~166bp的倍数——单核小体dna的长度(~146bp),并带有一些另外的连接体dna。健康人的血浆包含~1000个基因组/ml的等同量,并且上至100倍多的cfdna存在于很多病理情况(例如癌症)和一些生理状况(例如运动后)中。这些片段寿命短,并且估测半衰期短于1小时,使其成为以无创方式监测生理和病理过程的理想生物标记。近年来,cfdna作为诊断工具的使用已大幅扩展。例如,现在将母体血液中胎儿cfdna的下一代测序技术用于无创的染色体异常和亲本源突变的产前筛查/诊断。由于血液样品中存在的cfdna量非常低,大多数当前的cfdna诊断方法都依赖于cfdna的突变来将其与健康组织和血细胞中的cfdna区分。事实上,到目前为止,血细胞cfdna是cfdna总库的最大贡献者,并且可使其他组织的状况的cfdna诊断变得困难。
大多数当前基于cfdna的方法都依赖于检测cfdna中的基因组变化,以定量基因组序列(如胎儿、移植物、或肿瘤突变基因)改变的细胞的cfdna的贡献。因此,这些方法偏重一组预选的基因,而无视涉及基因组与宿主基因组相同的细胞的周转(turnover)和死亡的事件。最近的方法利用了无细胞dna中的表观遗传信息。总cfdna的极深测序可提供反映来源组织和基因表达的数据。但是,其依赖于检测目标区域覆盖率的变化,其中源组织的信号被强加在正常细胞的背景上(例如,在10%的细胞中导致核小体耗竭的事件的检测需要区分90%占据与100%占据)。因此,这种方法通过利用极深的测序覆盖(每个样品几亿个读段)来避免采样噪声。即使有这样的测序深度,对于稀有亚组的细胞中的事件也存在严格的苛刻检测限制。一种有前景的替代方法是测定沿着序列的dnacpg甲基化以鉴定来源细胞。dna甲基化充当稳定的表观遗传记忆,并且在分化后大部分保持不变。因此,其关于细胞谱系提供很多信息,但关于表达的瞬时变化以及源自相近或相似谱系的细胞却很少。此外,dna甲基化的无偏分析需要高测序深度,因为大多数cpg都是甲基化的。
允许准确确定cfdna来源并提供关于细胞中接近细胞死亡时发生的分子事件的信息的方法将不仅允许对医生或患者未知的状况的早期诊断,而且还可有助于根据新发现疾病调整治疗。
技术实现要素:
本发明提供了确定无细胞dna(cfdna)的来源、检测细胞类型或组织的死亡、确定对象中的细胞的细胞状态及其组合的方法,其通过对通过提取蛋白质以及与cfdna结合的修饰蛋白质分离出的该cfdna测序而进行。还提供了用于这样做的计算机程序产品。
根据第一方面,提供了确定释放其dna的细胞的细胞状态、来源组织、细胞类型或其组合的方法,包括:
a.提供样品,其中所述样品包含无细胞dna(cfdna);
b.使样品与至少一种与dna缔合蛋白结合的试剂接触;
c.分离试剂和任何其结合的蛋白质和cfdna;
d.对分离的cfdna测序;和
e.指定包含信息性(informative)基因组位置的dna序列的cfdna分子源自处于某细胞状态的细胞,源自某组织,源自某细胞类型,或其组合,其中dna缔合蛋白与信息性基因组位置的缔合指示释放该cfdna的细胞的细胞状态、来源组织、细胞类型或其组合;从而确定释放其dna的细胞的细胞状态、来源组织、细胞类型或其组合。
根据另一方面,提供了用于确定无细胞dna(cfdna)的来源细胞或组织的计算机程序产品,其包括非暂时性计算机可读存储介质,该非暂时性计算机可读存储介质上包含程序代码,该程序代码可被至少一个硬件处理器执行以:
a.测量或访问(access)用结合dna缔合蛋白的试剂分离的cfdna的测序;
b.将来自cfdna的cfdna分子分配给某来源细胞或组织,所述分配通过将分子的dna序列和所述细胞类型或组织中与所述dna缔合蛋白缔合的序列比较进行;和
c.提供关于cfdna的来源细胞或组织的输出。
根据另一方面,提供了用于确定对象中的细胞在该细胞死亡时的细胞状态、来源组织、细胞类型或其组合的计算机程序产品,其包括非暂时性计算机可读存储介质,该非暂时性计算机可读存储介质上包含程序代码,该程序代码可被至少一个硬件处理器执行以
a.测量或访问用结合dna缔合蛋白的试剂分离的来自对象的cfdna的测序;
b.将cfdna中的cfdna分子分配给某细胞状态、来源组织、细胞类型或其组合,所述分配通过将所述分子的dna序列和所述细胞状态、组织、细胞类型或其组合中与所述dna缔合蛋白缔合的序列比较进行;和
c.提供关于所述对象中的细胞在所述细胞死亡时的细胞状态、来源组织、细胞类型或其组合的输出。
根据另一方面,提供了固体载体,其包含捕获剂和条形码化试剂。
根据另一方面,提供了对单一溶液中的多于一种目标分子多重化(multiplexing)测定的方法,该方法包括:
a.在溶液中将第一目标分子捕获至本发明的第一固体载体;
b.在溶液中将至少第二目标分子捕获至本发明的第二固体载体;
c.将第一目标分子和第一条形码以及至少第二目标分子和第二条形码附接;
d.同时对第一和第二目标分子进行测定,其中第一目标分子的测定结果通过第一条形码识别,并且第二目标分子的测定结果通过第二条形码识别;
从而对单一溶液中的多于一种目标分子多重化测定。
根据一些实施方式,样品来自对象。
根据一些实施方式,释放其dna的细胞是死亡的细胞,并且该方法用于检测以下至少一种的死亡:
a.对象中的某细胞类型,
b.对象中的某组织,以及
c.在对象中处于某细胞状态的细胞。
根据一些实施方式,细胞状态是疾病状态。根据一些实施方式,疾病状态选自菌血症(bacteremia)、癌症、癌前(pre-cancer)、感染、神经变性疾病、组织损伤、心脏疾病、肝脏疾病、炎症、自身免疫疾病、关节炎、肝脏炎症、肠炎症、自身免疫疾病、由于药物副作用的组织损伤、组织坏死和糖尿病。根据一些实施方式,疾病状态选自心脏疾病或损伤、脑疾病或损伤、胃肠道疾病或损伤、癌症、菌血症、感染和肝脏疾病或损伤。
根据一些实施方式,提供了至少500个基因组的cfdna。根据一些实施方式,所述指定可以用样品中的来自所述细胞类型、所述组织或所述细胞状态的少至0.1%的cfdna进行。
根据一些实施方式,试剂选自抗体或其抗原结合片段、蛋白质或小分子。
根据一些实施方式,试剂被缀合至物理载体。
根据一些实施方式,dna缔合蛋白选自组蛋白、高迁移率族(hmg)蛋白和转录机制成员。根据一些实施方式,组蛋白是组蛋白变体和/或修饰组蛋白。根据一些实施方式,组蛋白变体选自组蛋白3单甲基化赖氨酸4(h3k4me1)、组蛋白3去甲基化赖氨酸4(h3k4me2)、组蛋白3三甲基化赖氨酸36(h3k36me3)和组蛋白3三甲基化赖氨酸4(h3k4me3)。根据一些实施方式,试剂是抗修饰组蛋白抗体或其片段。
根据一些实施方式,dna缔合蛋白与所述基因组位置的缔合指示活跃的(有活性的,active)转录,并且所述基因组位置在某组织、细胞类型或细胞状态的特异性基因或增强子元件内或处于疾病特异性的突变处。根据一些实施方式,dna缔合蛋白与所述基因组位置的缔合指示沉默的转录,并且所述基因组位置在阻遏元件、或者所述组织、细胞类型或细胞状态下沉默的基因内,或处于疾病特异性的突变处。
根据一些实施方式,本发明的方法进一步包括使用与第二dna缔合蛋白结合的试剂再次执行步骤a-d,并且其中第二dna缔合蛋白不同于第一dna缔合蛋白。
根据一些实施方式,本发明的方法包括使样品与至少2种试剂接触,其中各试剂结合至物理载体,并且载体包含各试剂独特的短dna标签,其中在对分离的cfdna测序后,所述短dna标签识别使cfdna分离的试剂。
根据一些实施方式,所述指定包括将测序的cfdna与某组织、细胞类型或细胞状态中具有dna缔合蛋白的最独特缔合的至少10个基因组位置比较,并且其中具有与所述至少10个基因组位置内的dna序列相同的序列的cfdna被认为来自所述组织、细胞类型或细胞状态。
根据一些实施方式,所述dna缔合蛋白是活跃转录的标记,并且所述指定包括将测序的cfdna与某组织、细胞类型或细胞状态的已知转录程序比较,其中具有来自所述转录程序中所转录的基因的序列的cfdna来自所述组织、细胞类型或细胞状态。
根据一些实施方式,所述指定包括将测序的cfdna与至少5种细胞类型或组织的dna-蛋白质缔合图谱(atlas)进行比较,其中所述图谱包含所述5种细胞类型或组织每一种中具有dna缔合蛋白的最独特缔合的至少10个基因组位置,并且其中具有与所述至少10个基因组位置内的dna序列相同的序列的cfdna被认为来自所述组织或细胞类型。
根据一些实施方式,所述指定包括将测序的cfdna与至少5种转录程序的转录程序图谱进行比较,其中所述图谱包含所述5种转录程序每一种中具有dna缔合蛋白的最独特缔合的至少一个基因组位置,并且其中具有与所述至少一个基因组位置内的dna序列相同的序列的cfdna指示转录程序的激活。
根据一些实施方式,细胞状态选自:缺氧、炎症、er应激、线粒体应激、干扰素响应、休眠、衰老、循环、恶性和钙流动。
根据一些实施方式,信息性基因组位置选自启动子、增强子元件、沉默子元件、基因体(genebody)和疾病相关突变。
根据一些实施方式,本发明的方法其中:
a.dna缔合蛋白是活跃转录的标志物,并且疾病相关突变处于癌基因内,或
b.dna缔合蛋白是沉默转录的标志物,并且疾病相关突变处于肿瘤抑制基因(tumorsuppressorgene)内。
根据一些实施方式,本发明的方法用于检测对象的疾病状态。
根据一些实施方式,本发明的方法其中检测疾病状态包括以下至少一种:
a.疾病状态的早期检测;
b.残余转移性疾病的检测;和
c.监测有无治疗情况下的疾病进展。
根据一些实施方式,本发明的方法进一步包括基于对象中死亡的细胞的细胞状态、来源组织、细胞类型或其组合,用合适的治疗来治疗对象。
根据一些实施方式,固体载体是磁珠或顺磁珠、或琼脂糖珠。
根据一些实施方式,捕获剂是蛋白质。根据一些实施方式,捕获蛋白是抗体或其抗原结合片段。
根据一些实施方式,条形码化试剂是短核酸分子。根据一些实施方式,该核酸分子为5至30个核苷酸。
根据一些实施方式,将捕获剂和条形码化试剂缀合至固体载体。
根据一些实施方式,目标分子是蛋白质或核酸分子。
根据一些实施方式,测定是染色质免疫沉淀,然后测序(chip-seq)。
本发明的其他实施方式和可应用的全部范围将由下文给出的详细描述中而显而易见。然而,应该理解,详细描述和具体实例在指出本发明的优选实施方式时仅是通过示例的方式给出的,因为本发明的精神和范围内的各种改变和修改对于本领域技术人员而言将是由该详细描述显而易见的。
附图说明
本文参考附图描述了本发明的一些实施方式仅作为实例。在现具体地详细参考附图时,注意所示出的细节是作为实例并且以对本发明的实施方式的示例性讨论为目的。就这点而言,说明书和附图使得如何可以实践本发明的实施方式对于本领域技术人员而言显而易见。
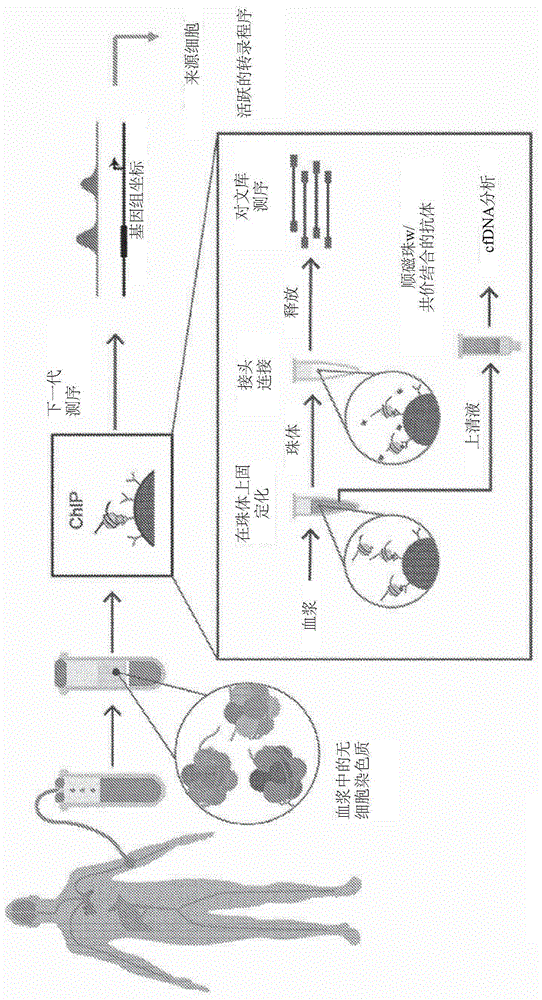
图1a-1i:(1a)所提出方法的概述。来自体内不同细胞的染色质片段被释放到血液中。将这些免疫沉淀并测序。对所得序列的解释告知了来源组织和基因活性程序。inset-cfchip方案,使用与顺磁珠共价结合的抗体。直接从血浆中免疫沉淀目标片段。除去血浆并洗涤结合到目标片段的珠体后,使用珠上连接(on-bead-ligation)将测序接头(可能带有索引条形码)添加到片段,并分离连接的dna,和进行pcr扩增,测序就绪文库准备就绪。(1b)从roadmapepigenomics数据汇编的细胞类型特异性的h3k4me1和h3k4me3位点的读段(reads)热图。显示了单个组织/细胞类型和/或相关细胞群的特异性位点。(1c)2号染色体的比对区段,显示cfchip-seq信号。上轨迹是来自四个被确定健康的对象的cfchip-seq信号。下轨迹是人类白细胞(白细胞)和组织的已公开chip-seq结果。下方是100倍放大的结果,显示了峰位置的一致性。(1d)活性启动子和增强子上的cfchip信号的元分析(荟萃分析,meta-analysis)的直方图。(ie)测序cfchip片段的尺寸分布直方图显示清晰的单核小体和双核小体尺寸。(if)出现在健康对象中但未出现在血细胞和实体组织的chip中的具有巨核细胞特异性基因的两个区域上的cfchip-seq信号的浏览器视图。(1g)在选定基因的启动子处的非pbmch3k4me3信号的浏览器视图(类似于图1f)。上图和下图分别描绘了cfchip和组织chip信号。(1h)在已知ctcf位点处的小鼠ctcf信号的浏览器视图。通过h3k4me3信号的耗尽来证实该位点。(1i)整个小鼠基因组中的小鼠ctcf(上)和h3k4me3(下)信号的元分析。
图2a-m:(2a-2b)比较cfchip与(2a)技术复制品和(2b)1位男性和1位女性健康个体的抗h3k4me1和抗h3k4me3抗体的散点图。各点是基因组中的一个2kb窗口,x和y轴是映射到所述两个样品中的窗口的读段数目(以log(x+1)比例)。颜色代码反映点的密度。(2c)健康对象的h3k4me3cfchip样品之间的相关性的直方图。(左)2kb窗口中计数的相关性(如图2a-b)和(右)基因启动子中计数的相关性。我们看到,来自相同对象的样品(红色直方图)倾向于比不同对象的样品(蓝色直方图)彼此之间的相关性略高。(2d)性别特异性峰的浏览器实例。将男性和女性血浆样品以已知比例混合,并进行h3k4me3的cfchip。(2e)2d显示的样品中男性特异性chry特征(signature)的检测的条形图。显示了背景信号的fdr调整q值。(2f)显示h3k4me3男性信号与分数(fraction)呈线性关系的图。比较基于100%男性样品和poisson样品的模拟中的读段计数vs观察到的数目。(2g)检出具有不同读段数目的特定位置的概率的线图估测。通过从实际结果中进行下采样(down-sampling)而估测检出概率。条形代表估测值的95%置信区间。(2h)较大特征尺寸的加标(spikein)的线图外推。显示了在两个样品量中检测到0.1%男性的概率。(2i)组织高度特异性特征的尺寸的条形图(见表1)。(2j)组成性表达基因启动子处的h3k4me3与rna水平之间的相关性的散点图。(上)pbmc(白细胞)的chip-seqvspbmc的rna-seq。(下)健康对象的cfchipvspbmc的rna-seq。(2k)h3k4me3cfchip-seq与表达水平的散点比较。各点是基因。x轴:基因启动子中的h3k4me3读段数目(标准化后;方法)。y轴:基因的白细胞rna-seq计数。(2l)健康对象的cfchip中检测到的组织特异性特征的点图。显示了其cfdna预期在cfdna中表达的细胞的特征计数:嗜中性粒细胞,35%cfdna;单核细胞,25%cfdna;和肝细胞,1%;以及阴性对照(心脏)的特征计数。各列中的点是特定对象的计数。(2m)显示图2l中特征的显著性的点图。
图3a-j:(3a)四个健康对象和来自心肌梗塞(mi)患者的样品的心脏特异性窗口中的h3k4me3cfchip-seq信号的条形图。插图,在抽取血液样品时测得的肌钙蛋白水平。(3b)心脏特异性窗口处的信号浏览器视图实例。各浏览器部分显示窗口周围20kb区域(标以灰色背景)。轨迹均已标准化,并以相同比例显示。上轨迹显示cfchip样品,并且下部组织样品(下)的chip-seq来自roadmap表观基因组学图谱(roadmapepigenomicsatlas)。(3c)与心肌细胞死亡的外部迹象的点图比较。x轴:测量的肌钙蛋白水平(上图),使用dna甲基化标志物测量的心肌细胞分数(下图)。y轴:心脏特异性特征的强度(相对于健康对象)。(3d)显示健康对象和心肌梗塞患者中所选细胞类型特征的水平(棕色标度)和显著性(蓝色标度)的热图。图中的各单元格分为两半,左上半部分代表统计学显著性(fdr校正的q值),下半部分代表特征中的读段密度(标准化读段/kb)。(3e)所有样品的组织特征的热图;以及3d和3i的扩展。(3f)作为被评价特征(见图3b)的部分的肝脏特异性窗口的浏览器视图实例。(3g)在pci之前/之后的心肌梗塞患者的特征强度变化的线图。特征强度被相对于健康对象标准化。健康对象之间的差异显示在左侧。我们可以看到最初高水平的肝细胞和升高水平的心脏细胞。随后pci肝细胞减少而心脏细胞增加。(3h)癌症患者的外部适应症和肝脏特征的点图比较。如图3c中显示。(3i)显示癌症患者的细胞类型特征(见图3d和3e)的热图。(3j)来自经历肝切除术的患者的血液样品的肝特征(条形)和alt水平(肝损害生物标志物,黑线)变化的组合线图和条形图。
图4a-h:(4a)显示对象中过表达(over-represented)的过程标志(hallmark)基因(与健康基线相比)的热图(如图3d)。见图4c,所有标志和对象的完全表格。(4b)这些表达特征中具有高于预期信号的基因的浏览器视图的实例(参见图3b)。(4c)所有样品和特征的标志特征的热图。扩展图4a。(4d)选定的糖酵解基因的启动子处的h3k4me3cfchip和组织chip信号的浏览器视图(见图4b)。(4e)用于限定特定样品中具有升高的信号的基因的方法的示例性散点图。各基因的启动子处的标准化h3k4me3计数的散点图。x轴:参考健康样品的平均值。y轴:在论(inquestion)样品中的计数。彩色点代表癌症特征中的基因。较大的点是显著过表达。(4f)显示在过表达基因中肿瘤特异性特征的富集的热图。各单元格被分为两半,左上半部分代表统计学显著性(fdr校正的q值),下半部分表示与特征的重叠(特征中基因数目百分比)。见图4g,所有肿瘤和对象的完全表格。(4g)所有样品的癌症特征的热图。(4h)不同样品中癌症相关基因及其信号的浏览器视图的实例。
图5a-m:(5a)活性启动子和增强子上的cfchip信号的元分析的直方图。(5b-c)来自健康对象的h3k4me3、h3k4me2和h3k4me13的cfchip轨迹的浏览器视图。(5b)显示了高表达基因的区域。我们可以看到二甲基化和单甲基化从三甲基化信号延伸出来。(5c)显示了ifnb1周围的基因座。chromhmm轨迹显示根据组蛋白修饰和染色质可及性测定的组合的启动子和增强子的预测。箭头标注了二甲基化和单甲基化富集的区域。(5d-e)显示(5d)健康对象和癌症患者两个样品中启动子处h3k4me2和h3k4me3的相关性以及(5e)健康对象的h3k4me2之间的一致性以及从癌症患者间隔数月采集的两个样品的h3k4me2之间的一致性的散点图。健康样品和癌症样品之间的差异显著。(5f-h)比较健康样品与癌症样品(c002.2)之间的h3k4甲基化标志物的轨迹浏览器视图,(5f)tcf3,(5g)cdx1,(5h)ceacam5和ceacam6。(5i)cfchip的侧接转录起始位点(tss)和转录终止位点(tes)的5kb的基因体上的h3k36me3信号的元分析的直方图。基因长度成比例。(5j)白细胞与健康样品之间h3k36me3相关性的散点图。(5k)h3k36me3的箱形图标注了活性基因-健康样品h3k36me3计数(按基因长度标准化),其被白细胞rna水平分位数打断。(5l)基因体处原始hf3k36me3计数的散点图。每个点代表一个基因。x轴:健康样品。y轴:结肠直肠腺癌样品。彩色点表示在结肠直肠腺癌(coad-红色)或多形性成胶质细胞瘤(gbm-绿色)中过表达的基因。(5m)显示健康对象与结肠直肠腺癌患者之间这些标志物的差异水平的基因处h3k4me3和h3k36me3信号的浏览器视图。vil1基因显示两个标志物的差异信号,而ctdsp1显示结肠直肠腺癌患者样品中h3k4me3的水平相似,但h3k36me3显著增加。
图6a-c:(6a-c)背景估测的线图实例,(6a)健康男性样品,(6b)健康女性样品,和(6c)癌症患者。
图7:cf-chip的处理和分析的工作流程。
图8:1000种高表达启动子的元(meta)作图(上)和热图(下)以及来自已经历利用抗h3k4me1抗体进行的cfchip处理的血浆的cf-核小体中的h3k4me3相对于转录起始位点(tss)的位置。
图9:1000种高表达启动子的元(meta)作图(上)和热图(下)以及健康患者和一名结肠直肠癌患者的血浆的cf-核小体中的h3k9ac、h3k27ac和h2a.zac相对于tss的位置。
图10a-d:(10a)描述多重化chip-seq的方案的示意图。(10b)在基于mpl的chip-seq过程中为测试混合所进行的实验示意图。每个矩形代表一个mpl条形码表面,其组合了独特的条形码(bc1-bc4)和抗h3k4me3(k4)或抗h3k36me3(k36)抗体的组合,各抗体靶向不同基因组位置的染色质修饰。椭圆是来自如下两个酵母物种的染色质:酿酒酵母(s.cerevisiae)和乳酸克鲁维酵母(k.lactis)。然后在文库制备(中间为粉色圆形)之前或之后进行各种混合。(10c)捕获的固定化染色质的分数的条形图(以输入量的%显示)。(10d)根据来自mpl条形码化表面的chlp-seq信号的h3k4me3和h3k36me3在基因体上的分布的线图元分析。
具体实施方式
本发明提供了确定无细胞dna(cfdna)的来源的方法、检测对象中细胞类型或组织的死亡的方法、以及通过由对象的有转录活性的或无转录活性的染色质确定dna-蛋白质缔合来确定细胞在其死亡时的细胞状态的方法。本发明的方法基于以下惊人的发现:无细胞核小体保留蛋白质-dna缔合,该蛋白质-dna缔合不仅关于核小体来源的组织/细胞具有信息性,而且关于细胞中在其死亡时有活性和无活性的途径具有信息性。进一步,即使可以捕获的cf-核小体的数量非常小,这也是惊人地可行。相当于少至一千个基因组的cf-核小体足以执行本发明的方法。
本发明的方法可以以少至1000个基因组的非常少的输入cfdna以及少至0.5m个读段的非常浅表的测序来进行。该技术可以如此执行是因为仅考察阳性缔合。不对完整cfdna进行测序,而仅分离和测序被具体蛋白质(例如修饰的组蛋白)结合的cfdna。由于仅对小部分cfdna测序,该过程较便宜,较快速,并且可以以较低测序深度完成。即使在这种较小的样品中,也仅考察信息性基因组基因座;大多数位置没有关于来源组织/细胞或死亡细胞中的活性途径的信息。通过仅考察信息性基因座,cfdna中存在的很多噪声可以被忽略。最后,由于研究在某些情况下仅与目标蛋白质缔合的dna序列,仅需这些区域中的少量读段即可鉴定cfdna中的阳性读段。例如,如果研究目标蛋白质与心脏组织中独特结合的dna序列的结合,则健康个人(没有或几乎没有心脏细胞死亡者)在这些区域中将只有屈指可数的读段(见图3a-e)。健康对象的被发现的组织和读段差异非常小,因此即使以很少的与健康个体不同的读段,也可以进行异常细胞死亡的检测。在心脏组织独特的基因组区域处具有增加的读段数目的对象将被鉴定为具有正在升高的心脏细胞死亡。无需测量这些区域内的每一个读段,因为阴性数据是无关的,仅超过健康个人的基线显著升高的读段就足够。这也可以被进行以研究垂死细胞的途径和细胞状态。由于来自健康对象的cfdna有很少的读段显示缺氧途径中的基因激活,在这些区域内的读段将指示缺氧是患者细胞死亡增加的原因。
cfchip有可能规避当前cfdna分析中存在的许多限制。活性标志物的靶向富集导致基因组表达(representation)减少,因此需要较少的测序读段(少~2个数量级)获得信息性信号。由于我们靶向与活跃转录相关的标志物,我们要分析阳性信号,其中很少的读段指示具体细胞类型或表达程序的存在。这与诸如占有率或dna甲基化的测量阴性信号(无核小体占有)或同时测量阴性信号和阳性信号(例如%甲基化)的方法形成对比。此外,cfchip测定可使大部分原始样品保持完整,能够将相同的材料用于多个测定(例如基因组测序、甲基化分析或其他抗体的cfchip),这在血量是限制因素的情况下是重要的。
在过去的二十年中,深入的研究建立了特定的组蛋白标志物与染色质模板化过程(包括转录、复制和损伤修复)之间的联系。利用这种丰富而复杂的信息进行血液cfdna分析,有可能实时并且以最小的侵入性揭示远程器官中的生理过程,例如细胞增殖、缺氧、炎症、代谢变化和癌性转化。这些过程全部都涉及大型转录程序的激活,这在染色质上留下独特的印记。
使用基于cfdna的测定来检测稀有细胞中的cfdna(如在早期癌症诊断中)的关键因素是低的检测限。cfchip的若干特征可以大大提高检测限。1.cfchip检测到“阳性”信号,因此即使低信号也有显著贡献。2.cfchip可以使用靶向不同基因组区域和状态的各种抗体来进行,从而产生大的特征以及在不同组织或转录程序之间具有差异信号的数百个或数千个位点的范围。3.由于cfchip本质上是低表达方法,因此cfchip是无偏见的,因为所有捕获的dna片段均被测序。
测定修饰的cf-核小体——单独应用或与现有生物标志物组合应用——具有多种潜在的医学用途,例如早期疾病检测(例如,检测未知肿瘤)、改善诊断(例如,用液体活检代替组织活检)和疾病进展和治疗功效的非侵入式监测。
第一方面,提供了确定无细胞dna(cfdna)的来源的细胞或组织的方法,该方法包括:
a.提供包含cfdna的样品;
b.使样品与结合dna缔合蛋白的至少一种试剂接触;
c.分离该试剂及其任何结合的蛋白质和cfdna;和
d.对分离的cfdna进行测序;
其中分离的cfdna包含某信息性基因组位置的dna序列,并且dna缔合蛋白与该信息性基因组位置的缔合指示细胞类型或组织;从而确定该cfdna的来源细胞或组织。
另一方面,提供了确定释放其dna的细胞的细胞状态、来源组织、细胞类型或其组合的方法,包括:
a.提供样品,其中所述样品包含无细胞dna(cfdna);
b.使样品与结合dna缔合蛋白的至少一种试剂接触;
c.分离该试剂及其任何结合的蛋白质和cfdna;
d.对分离的cfdna进行测序;和
e.指定包含某信息性基因组位置的dna序列的cfdna分子为来源于处于某细胞状态的细胞、来源于某组织、来源于某细胞类型或其组合,其中dna缔合蛋白与该信息性基因组位置的缔合指示释放该cfdna的细胞的细胞状态、来源组织、细胞类型或其组合;
从而确定释放其dna的细胞的细胞状态、来源组织、细胞类型或其组合。
另一方面,提供了确定无细胞dna(cfdna)来源的细胞或组织的方法,包括:
a.提供包含cfdna的样品;
b.使样品与结合dna缔合蛋白的至少一种试剂接触;
c.分离该试剂及其任何结合的蛋白质和cfdna;
d.对分离的cfdna进行测序;和
e.指定包含某信息性基因组位置的dna序列的cfdna分子为来源于某细胞类型或组织,其中dna缔合蛋白与该信息性基因组位置的缔合指示细胞类型或组织;
从而确定cfdna的来源细胞或组织。
另一方面,提供了确定无细胞dna的来源细胞或组织的方法,包括对通过cfdna与dna缔合蛋白结合而分离的cfdna进行测序;其中分离的cfdna包含某信息性基因组位置的dna序列,并且dna缔合蛋白与该信息性基因组位置的缔合指示细胞类型或组织;从而确定cfdna的来源细胞或组织。
另一方面,提供了确定对象中的细胞的细胞状态的方法,包括:
a.提供来自对象的样品,其中所述样品包含cfdna;
b.使cfdna与结合dna缔合蛋白的至少一种试剂接触;
c.分离该试剂及其任何结合的蛋白质和cfdna;和
d.对分离的cfdna进行测序;
其中分离的cfdna包含某信息性基因组位置的dna序列,并且dna缔合蛋白与该信息性基因组位置的缔合指示细胞状态;从而确定对象中的细胞的细胞状态。
另一方面,提供了确定对象中的细胞在该细胞死亡时的细胞状态的方法,包括:
a.提供来自对象的样品,其中所述样品包含cfdna;
b.使样品与结合dna缔合蛋白的试剂接触;
c.分离该试剂及其任何结合的蛋白质和cfdna;
d.对分离的cfdna进行测序;和
e.指定包含某信息性基因组位置的dna序列的cfdna分子为源自处于某细胞状态的细胞,其中dna缔合蛋白与该信息性基因组位置的缔合指示细胞状态;
从而确定对象中的细胞在该细胞死亡时的细胞状态。
另一方面,提供了确定对象中的细胞的细胞状态的方法,包括对通过cfdna与dna缔合蛋白的结合而分离的cfdna进行测序;其中分离的cfdna包含某信息性基因组位置的dna序列,并且dna缔合蛋白与该信息性基因组位置的缔合指示细胞状态;从而确定对象中的细胞的细胞状态。在一些实施方式中,细胞是对象中的已经死亡的细胞。
另一方面,提供了确定对象中的细胞在该细胞死亡时的细胞状态、来源组织或细胞类型的方法,包括:
a.提供来自对象的样品,其中所述样品包含cfdna;
b.使样品与结合dna缔合蛋白的试剂接触;
c.分离该试剂及其任何结合的蛋白质和cfdna;和
d.对分离的cfdna进行测序;
其中分离的cfdna包含dna缔合蛋白的组织或细胞类型特异性结合位点的dna序列,该组织或细胞类型特异性结合位点指示细胞类型或来源组织,并且dna缔合蛋白与该组织或细胞类型特异性结合位点的缔合指示细胞状态;从而确定对象中的细胞的细胞状态。
另一方面,提供了确定对象中的细胞在该细胞死亡时的细胞状态、来源组织或细胞类型的方法,包括:
a.提供来自对象的样品,其中所述样品包含cfdna;
b.使样品与结合dna缔合蛋白的试剂接触;
c.分离该试剂及其任何结合的蛋白质和cfdna;
d.对分离的cfdna进行测序;和
e.将包含dna缔合蛋白的某组织或细胞类型特异性结合位点的dna序列的cfdna分子指定为源自该组织或细胞类型,以及源自处于某细胞状态的细胞,其中dna-缔合蛋白与结合位点的缔合指示细胞状态;
从而确定对象中的细胞在该细胞死亡时的细胞状态和来源组织或细胞类型。
在一些实施方式中,该方法用于确定细胞的细胞状态。在一些实施方式中,该方法用于确定细胞的来源组织。在一些实施方式中,该方法用于确定细胞的细胞类型。在一些实施方式中,样品来自对象,并且该方法用于检测对象中的某组织的细胞、某细胞类型和处于某细胞状态的细胞中的任一种的死亡。在一些实施方式中,样品来自对象,并且该方法用于检测对象中的疾病,其中某组织的细胞、某细胞类型的细胞或某细胞状态的细胞的死亡指示该疾病。关于非限制性实例,肝细胞的死亡可以指示肝脏疾病,gi细胞的死亡可以指示gi癌症,具有活跃干扰素响应的细胞的死亡可以指示感染,并且β细胞的死亡可以指示胰腺损伤/疾病。
在一些实施方式中,检测疾病状态包括以下至少一项:疾病状态的早期检测、残余疾病的检测和监测疾病进展。在一些实施方式中,检测疾病状态包括早期检测。在一些实施方式中,早期检测包括在常规血液工作期间的检测。在一些实施方式中,早期检测包括在症状发展之前进行检测。在一些实施方式中,残余疾病是残余转移性疾病。在一些实施方式中,残余疾病是手术后的残余癌症。在一些实施方式中,疾病监测包括治疗前的监测。在一些实施方式中,疾病监测包括治疗后的监测。在一些实施方式中,疾病监测包括监测疾病复发。在一些实施方式中,疾病监测包括监测治疗功效。
在一些实施方式中,细胞已死亡。在一些实施方式中,细胞释放其dna。在一些实施方式中,释放其dna的细胞是死亡和/或垂死的细胞或去核的细胞。在一些实施方式中,释放其dna的细胞是死亡和/或垂死的细胞。在一些实施方式中,细胞死亡选自凋亡死亡和坏死性死亡。在一些实施方式中,去核的细胞是红细胞。在一些实施方式中,失去其核的细胞是成红细胞。成红细胞失去其核而变成红细胞,因此,失去的核可存在于cfdna中。
在一些实施方式中,样品来自对象。在一些实施方式中,cfdna来自对象,并且检测到某细胞来源组织或细胞状态的cfdna分子指示检测到该细胞类型、组织或细胞状态的死亡。在一些实施方式中,对象疑似具有增加的细胞死亡。在一些实施方式中,对象未疑似具有增加的细胞死亡。在一些实施方式中,对象看起来是健康的和/或未知患有某疾病或状况。
在一些实施方式中,所述确定是确定细胞在其死亡时的细胞状态。在一些实施方式中,本发明的方法进一步用于确定对象中的细胞在该细胞死亡时的细胞状态,并且进一步包括指定包含某信息性基因组位置的dna序列的cfdna分子为源自处于某细胞状态的细胞,其中dna缔合蛋白与该信息性基因组位置的缔合指示该细胞状态。
在一些实施方式中,样品是体液。在一些实施方式中,体液是血液。在一些实施方式中,体液选自:血液、血清、胃液、肠液、唾液、胆汁、肿瘤液、脑脊液、母乳、精液、尿液、阴道液、间质液和粪便中的至少一种。用于无细胞dna提取的标准技术是技术人员已知的,其非限制性实例是qiaamp循环核酸试剂盒(qiagen)。
如本文所用,“结合……的试剂”是指任何蛋白质结合分子或组合物。蛋白结合是本领域公知的,并且可以通过本领域已知的任何测定法进行评估,包括但不限于酵母-双-杂交(yeast-2-hybrid)、免疫沉淀、竞争测定法、噬菌体展示、串联亲和纯化和邻近连接测定法。在一些实施方式中,试剂是蛋白质分子。在一些实施方式中,试剂选自抗体或其抗原结合片段、蛋白质和小分子。结合特定蛋白质的小分子是本领域公知的,并且可以用于下拉(pull-down)实验。另外,充分表征的蛋白质-蛋白质相互作用可用于下拉(实验)。事实上,可以用于沉淀、免疫沉淀(ip)或染色质免疫沉淀(chip)的任何试剂都可以用作该试剂。在一些实施方式中,试剂是抗体或其抗原结合片段。
如本文所用,术语“抗体”是指包括至少一个结合结构域的多肽或多肽组,所述结合结构域由具有三维结合空间的多肽链折叠形成,其中内部表面形状和电荷分布与抗原的抗原决定簇的特征互补。抗体通常具有四聚体形式,包含两对相同多肽链,每对具有一条“轻”链和一条“重”链。各轻/重链对的可变区形成抗体结合位点。抗体可以是寡克隆抗体、多克隆抗体、单克隆抗体、嵌合抗体、骆驼化抗体、cdr嫁接抗体、多特异性抗体、双特异性抗体、催化性抗体、人源化抗体、完全人抗体、抗独特型抗体以及可以以可溶形式或结合形式标记的抗体以及片段——包括表位结合片段、其变体或衍生物,单独或与其他氨基酸序列组合。抗体可以来自任何物种。术语抗体还包括结合片段,包括但不限于fv、fab、fab'、f(ab')2单链抗体(svfc)、二聚体可变区(双抗体)和二硫键连接的可变区(dsfv)。具体地,抗体包括免疫球蛋白分子和免疫球蛋白分子的免疫学活性片段,即含有抗原结合位点的分子。抗体片段可以融合或可以不融合至另一免疫球蛋白结构域,所述另一个免疫球蛋白结构域包括但不限于fc区或其片段。技术人员将进一步理解,可以生成其他融合产物,包括但不限于scfv-fc融合、可变区(例如vl和vh)-fc融合和scfv-scfv-fc融合。
免疫球蛋白分子可以是任何类型(例如igg1、ige、igm、igd、iga和igy)、类别(例如igg1、igg2、igg3、igg4、iga1和iga2)或亚类别的。
在一些实施方式中,接触一种试剂。在一些实施方式中,接触至少一种试剂。在一些实施方式中,接触多于一种试剂。在一些实施方式中,各试剂结合不同的dna缔合蛋白。在一些实施方式中,dna缔合蛋白是序列特异性的dna结合物,并且接触靶向多于一种蛋白质的多于一种试剂。由于结合的目标序列是已知的,在对分离的cfdna测序之后,可以基于序列中存在的目标基序将序列分配至各结合试剂。包含多个一种基序的序列可以被舍弃,或被被包括为被多种dna缔合蛋白质结合。
在一些实施方式中,试剂被缀合至物理载体。如本文所用,术语“物理载体”是指为试剂提供支持的固体和稳定分子。在一些实施方式中,载体是支架或支架剂。在一些实施方式中,载体是树脂。在一些实施方式中,载体是珠。在一些实施方式中,载体是磁珠或顺磁性珠。磁珠可以例如从dynabeads或pierce购买。在一些实施方式中,载体是琼脂糖珠。在一些实施方式中,载体是蛋白质a/g珠。在一些实施方式中,将试剂在接触之前缀合至物理载体。在一些实施方式中,缀合是共价键。在一些实施方式中,缀合通过环氧化学进行的。在一些实施方式中,载体有助于试剂的分离,其中所述分离是分离物理载体。
如本文所用,术语“dna缔合蛋白”是指可以与dna一起沉淀或在沉淀时连同dna获得的任何蛋白质。在一些实施方式中,dna缔合蛋白直接结合dna。在一些实施方式中,dna缔合蛋白是染色质的组分。在一些实施方式中,dna缔合蛋白间接结合dna。在一些实施方式中,dna缔合蛋白结合基因组dna。在一些实施方式中,dna结合蛋白结合在启动子中。在一些实施方式中,dna结合蛋白结合在基因体中。在一些实施方式中,dna结合蛋白结合顺式或反式调控元件。
在一些实施方式中,dna缔合蛋白结合dna,并且是非序列特异性的dna结合物。在一些实施方式中,dna缔合蛋白结合dna,是序列特异性dna结合物或非序列特异性dna结合物。非序列特异性dna结合物的实例包括组蛋白、高迁移率族(hmg)蛋白质、dna损伤修复机制的成员和一般转录机制的成员。一般转录机制是被明确地定义,并且包括但不限于rna聚合酶、dna解旋酶、一般辅因子、剪接机制和多聚a机制。dna损伤修复机制也被明确地定义,并且包括但不限于核苷酸切除修复途径、碱基切除修复途径和错配修复系统的成员。在一些实施方式中,dna缔合蛋白是修饰的蛋白质。在一些实施方式中,修饰是翻译后修饰。在一些实施方式中,试剂结合蛋白质的修饰形式。在一些实施方式中,试剂仅或主要结合蛋白质的修饰形式。
在一些实施方式中,dna缔合蛋白是组蛋白、修饰的组蛋白或组蛋白变体。组蛋白尾部的修饰在本领域公知的,包括但不限于甲基化、乙酰化、磺酰化、泛素化和磷酸化。修饰可以是多重的,例如三甲基化或多泛素化。在一些实施方式中,尾部可具有多重修饰,如甲基化和磷酸化。组蛋白可以是核心组蛋白h1、h2a、h2b、h3和h4中的一种,或者其可以是组蛋白变体,如,作为非限制性实例,h2a.z、γh2ax、h1t和h3.3。在一些实施方式中,修饰或变体组蛋白对转录具有激活功能或阻遏功能。在一些实施方式中,修饰型或变体有助于常染色质或异染色质的形成。在一些实施方式中,修饰组蛋白选自组蛋白3单甲基化赖氨酸4(h3k4me1)、组蛋白3二甲基化(去甲基化,demethylated)赖氨酸4(h3k4me2)、组蛋白3三甲基化赖氨酸36(h3k36me3)和组蛋白3三甲基化赖氨酸4(h3k4me3)。
在一些实施方式中,dna缔合蛋白结合dna,并且是序列特异性的dna结合物。序列特异性dna结合物的实例包括但不限于转录因子(tf)、活化剂、阻遏剂、隔离剂、dna修饰酶和一般转录机制的成员。在一些实施方式中,dna缔合蛋白是转录因子。在一些实施方式中,dna缔合蛋白是隔离剂。在一些实施方式中,转录因子选自活化剂、阻遏剂、隔离剂、dna修饰酶和一般转录机制的成员。在一些实施方式中,转录因子选自活化剂、阻遏剂和隔离剂。在一些实施方式中,转录因子是隔离剂。在一些实施方式中,转录因子是ctcf。
如本文所用,术语“转录因子”是指不是一般转录机制的部分但控制/调节dna序列的转录速率的任何蛋白质。在一些实施方式中,tf是在启动子区域中结合的因子。转录因子在本领域公知是与其结合的试剂。用tf进行chip也是公知的。
在一些实施方式中,该剂(agent)结合与组织和/或细胞类型特异性增强子元件结合的转录因子。在一些实施方式中,cfdna中的dna序列是位于组织和/或细胞类型特异性增强子元件处的序列。由于组织/细胞类型特异性,tf与cfdna中该元件的缔合表明cfdna来自该组织和/或细胞类型。由于增强子元件增强特定目标的转录,该目标可以指示细胞的细胞状态。在这种情况下,tf与基因组基因座的一种缔合可以提供来源组织/细胞和细胞状态的信息。其非限制性实例是组织特异性nf-κb增强子结合。已知nf-κb仅结合在各种组织(例如心脏)中的特定位点并介导炎症。因此,用抗nf-kb剂进行的分离以及然后cfdna中的组织特异性增强子序列的鉴定不仅指示cfdna的细胞来源,而且指示该细胞在死亡时处于炎性状态。在一些实施方式中,dna缔合蛋白是转录因子(tf),并且结合位点是tf结合位点。
如本文所用,“活化剂/激活剂”是指增加转录的蛋白质。在一些实施方式中,活化剂结合dna中的增强子元件。在一些实施方式中,活化剂与启动子近侧或远侧的元件结合。如本文所用,“阻遏剂”是指减少转录的蛋白质。在一些实施方式中,阻遏剂结合dna中的阻遏元件。在一些实施方式中,阻遏剂与启动子近侧或远侧的元件结合。
如本文所用,“隔离剂”是指隔离具有不同染色质结构或转录速率的dna区域的蛋白质。在一些实施方式中,隔离剂是增强子-阻断剂。在一些实施方式中,隔离剂隔离常染色质和异染色质。隔离剂的非限制性实例包括ctcf、gypsy和bdf1。在一些实施方式中,隔离剂结合在启动子和基因体的外侧。
dna修饰酶是本领域公知的,并且实例包括碱基/核苷酸切除修复机制、dna甲基转移酶和dna脱甲基酶的成员。
在一些实施方式中,dna缔合蛋白不结合dna。在一些实施方式中,蛋白质修饰结合dna的蛋白质。其实例包括但不限于组蛋白修饰酶和聚梳(polycomb)蛋白。
在一些实施方式中,dna缔合蛋白与信息性遗传基因座的缔合是组织或细胞类型特异性的。在一些实施方式中,dna缔合蛋白与信息性遗传基因座的缔合是分化特异性的。在一些实施方式中,dna缔合蛋白与信息性遗传基因座的缔合是细胞状态特异性的。在一些实施方式中,dna缔合蛋白与信息性遗传基因座的缔合指示转录激活、活跃转录、有转录活性的染色质或其组合。在一些实施方式中,dna缔合蛋白与信息性遗传基因座的缔合指示转录沉默、转录缺乏、无转录活性的染色质或其组合。转录不需在结合的遗传基因座处,而可以在近处或远处基因处,如激活剂和抑制剂的情况。如本文所用,术语“遗传基因座”和“基因组位置”同义,并且是指可以被蛋白质结合的dna具体区域。在一些实施方式中,遗传基因座是tf结合位点或dna的一些其他短序列。在一些实施方式中,基因座为2至20、2至16、2至12、2至10、2至8、2至6、2至4、4至20、4至16、4至12、4至10、4和8或4至6个碱基对。各可能性代表本发明的单独实施方式。在一些实施方式中,基因座是dna的核小体或核小体长度(~170bp)。在一些实施方式中,遗传基因座在150至190bp或160至180bp之间。
如本文所用,术语“信息性基因组位置”和“信息性遗传基因座”是同义使用的,并且是指基因组中具体位置的独特dna序列,该独特dna序列在与给定的dna缔合蛋白缔合时提供关于其中发生缔合的细胞的信息。在一些实施方式中,其提供关于其中发生缔合的细胞的来源组织或细胞类型的信息。在一些实施方式中,位置是组织或细胞类型特异性结合/缔合位点。在一些实施方式中,结合/缔合不是特异性的/独特的,而是在该组织或细胞类型中高度富集的。在一些实施方式中,其提供关于其中发生缔合的细胞的细胞状态的信息。在一些实施方式中,其提供关于其中发生缔合的细胞的来源组织和/或细胞类型以及细胞状态的信息。在一些实施方式中,其提供关于细胞中的疾病的信息。在一些实施方式中,其提供关于细胞中的转录程序的信息。
如本文所用,术语“转录程序”是指在转录方面协同作用的一组基因。该基因可以被活跃地转录和/或抑制,和/或是无活性的,和/或可及的,和/或不可及的。在一些实施方式中,该基因全部一起被转录调控。在一些实施方式中,转录程序指示细胞状态。在一些实施方式中,转录程序指示活跃的信号传导途径。组织特异性的、细胞类型特异性的、细胞状态特异性的和/或转录程序的特征可以在例如在roadmapepigenomics项目(roadmapepigenomics.org)、cancergenomeatlas(cancergenome.nih.gov)、genotype-tissueexpression(gtex)项目(gtexportal.org)或xena项目(xena.ucsc.edu)中找到。本文提供的表格也提供了这种特征。
在一些实施方式中,试剂结合组蛋白。在一些实施方式中,试剂是抗组蛋白抗体或其片段。在一些实施方式中,试剂结合修饰或变体组蛋白。在一些实施方式中,试剂是抗修饰组蛋白抗体或其片段。在一些实施方式中,试剂是抗变体组蛋白抗体或其片段。在一些实施方式中,试剂选自抗组蛋白3单甲基化赖氨酸4(h3k4me1)抗体和抗组蛋白3三甲基化赖氨酸4(h3k4me3)抗体。
在一些实施方式中,分离包括分离与试剂缀合的物理载体。在一些实施方式中,分离包括使试剂以及结合的蛋白质和dna与物理载体接触,然后分离物理载体。在一些实施方式中,本发明的方法包括chip。在一些实施方式中,分离包括chip。在一些实施方式中,分离包括洗涤步骤。
在一些实施方式中,测序包括对至少平均100、200、300、500或1000万个测序读段进行测序。各可能性代表本发明的单独实施方式。在一些实施方式中,测序包括对至少100、200、300、500或1000万个测序读段进行测序。各可能性代表本发明的单独实施方式。在一些实施方式中,扩增的cfdna包含少于100、200、300、500或1000万个测序读段。各可能性代表本发明的单独实施方式。在一些实施方式中,测序的深度为至多100、200、300、500或1000万个测序读段。各可能性代表本发明的单独实施方式。
在一些实施方式中,测序包括cfdna的pcr扩增。在一些实施方式中,扩增包括条形码或其他dna序列的连接。在一些实施方式中,扩增在cfdna仍与蛋白质缔合时进行。在一些实施方式中,扩增在cfdna仍与物理载体缔合时进行。在一些实施方式中,扩增在不使cfdna与试剂和/或载体解离的情况下进行。
在一些实施方式中,方法进一步包括将测序数据与dna结合蛋白的组织/细胞类型特异性数据进行比较,其中蛋白质与被某组织/细胞类型的该蛋白特异性结合的序列的结合表明cfdna来自该组织/细胞类型。组织/细胞类型特异性结合数据可以在诸如encode协会、nihepigenomeroadmap协会和genetranscriptionregulation数据库(仅举几例)的来源中找到。在一些实施方式中,基因组位置在某组织或细胞类型特异性基因或元件内。在一些实施方式中,蛋白质与活跃转录相关,并且基因组位置在某组织或细胞类型特异性基因或增强子元件内。其非限制性实例包括位于组织特异性基因中的h3k4me3或增强子中的h3k4me1。组织特异性基因的非限制性实例包括心脏细胞中的tnni3和mybpc3,以及肝细胞中的c8a和c8b。特定蛋白质的组织表达水平可以在多个网站上找到,例如uniprot数据库(www.uniprot.org)和gtex门户网站(www.gtexportal.org)。组织特异性基因表达和调控也可以在多个地方找到,最著名的是tiger数据库(bioinfo.wilmer.jhu.edu/tiger)和humanproteinatlas(www.proteinatlas.org)。在一些实施方式中,蛋白质与沉默转录相关,并且基因组位置在阻遏元件或所述组织或细胞类型中特异性沉默的基因内。组织特异性蛋白质-dna结合是本领域公知的,并且可以在本文上述资源中找到。任何信息性基因座结合可以用来确定cfdna的来源。
在一些实施方式中,将测序数据与在具体组织/细胞类型中的结合的至少最高(top)1、2、5、10、20、30、40、50、60、70、80、90或100个峰进行比较。各可能性代表本发明的单独实施方式。在一些实施方式中,仅研究一种具体的组织/细胞类型。在一些实施方式中,来自至少1、2、3、5、10、15、20、25、30、35、40、45或50个组织或细胞/类型的结合数据用于与测序数据进行比较。各可能性代表本发明的单独实施方式。
在一些实施方式中,本发明的方法进一步包括将测序的cfdna与某组织、细胞类型和/或细胞状态中与dna缔合蛋白缔合最多的至少1个基因组位置进行比较,并且其中具有与所述至少一个基因组位置内的dna序列相同的序列的cfdna被认为来自该组织、细胞类型和/或细胞状态。在一些实施方式中,所述至少一个基因组位置具有dna缔合蛋白的最大独特缔合。如本文所用,术语“独特缔合”是指唯一地或几乎排他地发生在某组织或细胞类型或细胞状态内的缔合。因此,例如,如果选择例如10个最独特位置,则应考察仅在某特定组织、细胞类型或状态下具有蛋白质结合的位置,具体地应选择具有最高结合的10个。如果没有所需数量的具有完全独特结合的位点,则应选择具有最独特结合的位点。可以采用最大独特性的任何确定(方式)。其实例包括但不限于具有结合的其他组织最少,以及目标组织与其他组织之间结合量差异最大。
在一些实施方式中,本发明的方法进一步包括将测序的cfdna与至少2种细胞类型和/或组织的dna-蛋白质缔合图谱(atlas)进行比较,其中图谱包含所述2种组织、细胞类型和/或细胞状态的每一种中dna缔合蛋白的缔合最大的至少1个基因组位置,并且其中具有与所述至少1个基因组位置内的dna序列相同的序列的cfdna被认为是来自该组织、细胞类型和/或细胞状态。在一些实施方式中,基因组位置具有dna缔合蛋白的最大独特缔合。在一些实施方式中,图谱具有至少1、2、3、5、7、10、15或20种细胞类型和/或组织。各可能性代表本发明的单独实施方式。在一些实施方式中,图谱包含每组织、细胞类型和/或细胞状态至少1、2、5、10、15、20、25、30、35、40、45、50、60、70、75、80、90或100个基因组位置。各可能性代表本发明的单独实施方式。
表1中可找到与dna缔合蛋白缔合时指示组织或细胞类型的基因组位置的实例。表1给出了h3k4me3组织信息性位置的示例性位置。在一些实施方式中,图谱包括表1中的全部或部分位置。在一些实施方式中,将cfdna的测序与表1进行比较。
表1:h3k4me3组织的特定位置
在一些实施方式中,通过与dna-蛋白质缔合图谱比较对来自对象的测序数据进行去卷积(deconvoluted)。通过这种方式,可以确定样品中不同组织、细胞类型和/或细胞状态对总cf核小体的贡献百分比。在一些实施方式中,去卷积给出仅信息性cf核小体的贡献百分比。
在一些实施方式中,cfdna和cf核小体不是通过与健康组织数据比较而分析,而是通过机器学习(machinelearning)而分析。机器学习是本领域公知的,并且通过对患有已知状况的患者进行本发明的方法进行,机器学习算法可以学习识别在分离出特定dna缔合蛋白时所提供的cfdna序列中的特定疾病状态和状况。在一些实施方式中,分析至少1、2、3、4、5、6、7、8、9、10、15或20个具有特定状况的对象——在该算法可以识别新对象中的该状况之前。
如本文所用,术语“细胞状态”是指在细胞中活跃的状况或细胞响应或途径。在一些实施方式中,细胞状态是导致和/或引起细胞死亡的细胞状况。在一些实施方式中,细胞状态是细胞在其即将死亡之前的状态,但不是该死亡的直接原因。在一些实施方式中,细胞状态是细胞中在其死亡时活跃或不活跃的途径。在一些实施方式中,细胞状态是细胞中在其死亡时活跃或不活跃的细胞响应。在一些实施方式中,细胞状态包括提供关于细胞死亡原因的信息的至少一种基因的表达。在一些实施方式中,细胞状态是疾病状态。在一些实施方式中,细胞状态包括细胞中在其死亡时提供关于活跃途径的信息的至少一种基因的表达。在一些实施方式中,来自途径的至少1、2、3、4或5个基因指示活跃途径。各可能性代表本发明的单独实施方式。信号传导途径是本领域公知的,并且用于确定各种途径的成员的在线资源可以通过查阅例如geneontology或thermofisherscientific找到。
在一些实施方式中,确定细胞状态包括确定在细胞中活跃的细胞途径。在一些实施方式中,确定细胞状态包括确定在细胞中活跃的转录程序。在一些实施方式中,确定细胞状态包括确定提供关于活性途径信息的至少一种基因的活跃转录。在一些实施方式中,确定来自某途径的至少1、2、3、4或5个基因。各可能性代表本发明的单独实施方式。在一些实施方式中,确定细胞状态包括确定dna缔合蛋白与调控途径的基因的至少1个基因组区域的缔合。在一些实施方式中,细胞状态是缺氧、炎症、er应激、线粒体应激、休眠,衰老、干扰素响应、循环、恶性和钙流动中的任一种。通过本发明的方法可以研究可由基因或一组基因的表达限定的任何细胞状态。
在一些实施方式中,本发明的方法进一步包括将测序的cfdna与细胞途径的激活过程中具有dna缔合蛋白的最大缔合的至少1个基因组位置进行比较,并且其中具有与所述至少1个基因组位置内的dna序列相同的序列的cfdna指示细胞途径的激活。在一些实施方式中,比较具有最独特缔合的基因组位置。
在一些实施方式中,本发明的方法进一步包括将测序的cfdna与至少2个细胞途径的途径图谱进行比较,其中所述图谱包含所述2个细胞途径的每一个中具有dna缔合蛋白的最大缔合的至少1个基因组位置,并且其中具有与所述至少1个基因组位置内的dna序列相同的序列的cfdna指示该细胞途径的激活。
在一些实施方式中,将测序的cfdna与至少1、2、3、5、10、15、20、25、30、35、40、45、50、60、70、80、90或100个基因组位置进行比较。各可能性代表本发明的单独实施方式。在一些实施方式中,将测序的cfdna与至少10个基因组位置进行比较。在一些实施方式中,将测序的cfdna与至少25个基因组位置进行比较。在一些实施方式中,所述指定包括将测序的dna与给定数目的基因组位置进行比较。
在一些实施方式中,图谱是至少1、2、3、5、10、10、15、20、25、30、35、40、45或50种细胞类型和/或组织的图谱。各可能性代表本发明的单独实施方式。在一些实施方式中,图谱是至少1、2、3、5、10、10、15、20、25、30、35、40、45或50个细胞途径的图谱。各可能性代表本发明的单独实施方式。在一些实施方式中,所述图谱包含至少1、2、3、5、10、15、20、25、30、35、40、45、50、60、70、80、90或100个基因组位置。各可能性代表本发明的单独实施方式。在一些实施方式中,图谱包含至少2个基因组位置。在一些实施方式中,图谱包含至少10个基因组位置。在一些实施方式中,所述图谱包含至少25个基因组位置。
在一些实施方式中,dna缔合蛋白与基因组位置的缔合指示活跃的转录,并且基因组位置在某组织、细胞类型或细胞状态特异性的基因、增强子元件内或处于疾病特异性突变处。在一些实施方式中,dna缔合蛋白与基因组位置的缔合指示活跃的转录,并且基因组位置在某组织、细胞类型或细胞状态特异性的基因、增强子元件内。在一些实施方式中,dna缔合蛋白与基因组位置的缔合指示活跃的转录,并且基因组位置处于疾病特异性突变处。在一些实施方式中,dna缔合蛋白是活跃转录的标志物,并且dna缔合蛋白与疾病相关突变的缔合指示疾病状态。在一些实施方式中,dna缔合蛋白与基因组位置的缔合指示活跃的转录,并且疾病相关突变在癌基因内。在一些实施方式中,dna缔合蛋白与基因组位置的缔合指示沉默的转录,并且基因组位置在阻遏元件或所述组织、细胞类型或细胞状态中沉默的基因内,或在疾病特异性突变处。在一些实施方式中,dna缔合蛋白与基因组位置的缔合指示沉默的转录,并且基因组位置在阻遏元件或组织、细胞类型或细胞状态中沉默的基因内。在一些实施方式中,dna缔合蛋白与基因组位置的缔合指示沉默的转录,并且基因组位置在疾病特异性突变处。在一些实施方式中,dna缔合蛋白与基因组位置的缔合指示沉默的转录,并且疾病相关突变在肿瘤抑制基因内。癌基因和肿瘤抑制基因是本领域公知的。癌基因的实例包括但不限于wnt、ras、myc和erk。肿瘤抑制基因的实例包括但不限于p53、ptch、nf1、p27kip1和apc。
如本文所用,“cfdna”是指从生物体获得的、存在于生物体中细胞外的任何dna。如本文所用,“cf核小体”是指cfdna和与cfdna结合和/或缔合的任何蛋白质。在一些实施方式中,cf核小体包含cfdna和cf组蛋白。在一些实施方式中,cfdna与dna缔合蛋白缔合。在一些实施方式中,cfdna不是裸露的。在一些实施方式中,cfdna作为cf核小体处于样品中。在一些实施方式中,cfdna没有被交联。在一些实施方式中,本发明的方法进一步包括在接触之前使cfdna和dna缔合蛋白交联。在一些实施方式中,方法不包括在接触之前使cfdna和dna缔合蛋白交联。
在一些实施方式中,cfdna是从生物体获得的、存在于生物体中囊泡外的dna。无细胞dna是本领域公知的,并且总体上是指在体液内自由漂浮的dna。该dna总体上不被包封在囊泡中,因此不考虑诸如通过外来体或其他囊泡转运体进行的转运中的dna。在一些实施方式中,cfdna是来自垂死和/或死亡细胞的dna。在细胞死亡时,dna总体上被片段化(裂解,fragmented),并在细胞溶解时从细胞中释放。但是,这种dna并不全部立即被移除或清除,因此可以留存在生物体内。来自死亡细胞的dna经常进入血流。在一些实施方式中,裂解的泡状染色质也被包括在cfdna内。
在一些实施方式中,cfdna是哺乳动物cfdna。在一些实施方式中,cfdna是人cfdna。在一些实施方式中,cfdna来自哺乳动物或人基因组。在一些实施方式中,cfdna是胎儿dna。在一些实施方式中,dna是病毒dna。在一些实施方式中,dna是细菌dna。在一些实施方式中,dna是真菌dna。在一些实施方式中,dna是寄生物dna。在一些实施方式中,dna来自病原体。在一些实施方式中,dna来自生活在健康对象中的生物体。在一些实施方式中,cfdna从体液中提取。在一些实施方式中,所述提供包括提供体液和从体液分离cfdna。在一些实施方式中,所述提供包括提供包含cfdna的体液和在体液中进行所述接触。在一些实施方式中,本发明的方法对少至0.00001、0.00005、0.0001、0.0005、0.001、0.005、0.01、0.05、0.1、0.5、1、1.5、2、2.5或3ml的体液进行。在一些实施方式中,本发明的方法对少至2ml的体液进行。各可能性代表本发明的单独实施方式。在一些实施方式中,本发明的方法以小于0.00001、0.00005、0.0001、0.0005、0.001、0.005、0.01、0.05、0.1、0.5、1、1.5、2、2.5、3、3.5、4、4.5或5ml体液进行。各可能性代表本发明的单独实施方式。在一些实施方式中,体液是血液。
在一些实施方式中,cfdna是胎儿无细胞dna(cffdna)。在一些实施方式中,方法用于无创胎儿监测。在一些实施方式中,对象是胎儿的母亲。在一些实施方式中,方法用于确定胎儿的细胞的细胞状态。在一些实施方式中,方法用于确定胎儿的疾病。在一些实施方式中,法用于确定胎儿的遗传异常。在一些实施方式中,方法用于确定胎儿中的细胞死亡的来源。
由于生物体中cfdna的半衰期短,其提供当时该生物体中发生的细胞死亡的快照(snapshot)。在一些实施方式中,本发明的方法检测距从对象采集样品的时间最近1分钟、2分钟、3分钟、4分钟、5分钟、10分钟、15分钟、20分钟、25分钟、30分钟、35分钟、40分钟、45分钟、50分钟、55分钟、1小时、2小时、3小时、6小时、12小时、18小时、1天、2天、3天、4天、5天、6天、1周、2周、3周或一个月内发生的细胞死亡。各可能性代表本发明的单独实施方式。在一些实施方式中,本发明的方法检测从对象采集样品前刻发生的细胞死亡。在一些实施方式中,本发明的方法进一步包括在所述提供之前从对象提取样品,其中样品包含cfdna。在一些实施方式中,本发明的方法进一步包括在从对象获取样品之后并且在将其进行接触之前将样品冷冻或保持在约4度。通过冷冻或保持样品低温(cold),本发明的方法仍可以检测在从对象采集样品前刻发生的细胞死亡。
在一些实施方式中,提供至少0.00001、0.00005、0.0001、0.0005、0.001、0.005、0.01、0.05、0.1、0.5、1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90或100ngcfdna。各可能性代表本发明的单独实施方式。在一些实施方式中,提供少至0.00001、0.00005、0.0001、0.0005、0.001、0.005、0.01、0.05、0.1、0.5、1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90或100ngcfdna。各可能性代表本发明的单独实施方式。在一些实施方式中,提供至少50ng。在一些实施方式中,提供少至50ng。在一些实施方式中,提供至少7ng。在一些实施方式中,提供少至7ng。在一些实施方式中,提供至少0.5ng。在一些实施方式中,提供少至0.5ng。在一些实施方式中,提供的cfdna为0.1至1000、0.1至900、0.1至800、0.1至700、0.1至600、0.1至500、0.1至400、0.1至300、0.1至250、0.1至200、0.1至150、0.1至100、0.1.至90、0.1至80、0.1至70、0.1至60、0.1至50、0.1至40、0.1至30或0.1至20ng、0.1至10ng、0.1至5ng、0.1至1ng、0.5至1000、0.5至900、0.5至800、0.5至700、0.5至600、0.5至500、0.5至400、0.5至300、0.5至250、05至200、0.5至150、0.5至100、0.5.至90、0.5至80、0.5至70、0.5至60、0.5至50、0.5至40、0.5至30或0.5至20ng,0.1至10ng、0.5至5ng、0.5至1ng、1至1000、1至900、1至800、1至700、1至600、1至500、1至400、1至300、1至250、1至200、1至150、1至100、1至90、1至80、1至70、1至60、1至50、1至40、1至30或1至20ng,10至1000、10至900、10至800、10至700、10至600、10至500、10至400、10至300、10至250、10至200、10至150、10至100、10至90、10至80、10至70、10至60、10至50、10至40、10至30或10至20ng。各可能性代表本发明的单独实施方式。在一些实施方式中,提供至多0.00001、0.00005、0.0001、0.0005、0.001、0.005、0.01、0.05、0.1、0.5、1、5、10、20、30、40、50、60、70、80、90、100、150、200、250、300、400、500、600、700、800、900或1000ngcfdna。各可能性代表本发明的单独实施方式。1000个基因组大约相当于6.6ngcfdna。
在一些实施方式中,cfdna包含至少0.00001、0.00005、0.0001、0.0005、0.001、0.005、0.01、0.05、0.1、0.5、1、2、5、10、50、100、200、300、500、700、800、1000、2000、3000、4000、5000、6000、7000、8000、9000或10000个基因组。各可能性代表本发明的单独实施方式。在一些实施方式中,cfdna包含少至0.00001、0.00005、0.0001、0.0005、0.001、0.005、0.01、0.05、0.1、0.5、1、2、5、10、50、100、200、300、500、700、800、1000、2000、3000、4000、5000、6000、7000、8000、9000或10000个基因组。各可能性代表本发明的单独实施方式。在一些实施方式中,cfdna包含0.1至10000、0.1至9000、0.1至8000、0.1至7000、0.1至6000、0.1至5000、0.1至4000、0.1至3000、0.1至2000、0.1至1000、1至10000、1至9000、1至8000、1至7000、1至6000、1至5000、1至4000、1至3000、1至2000、1至1000、5至10000、5至9000、5至8000、5至7000、5至6000、5至5000、5至4000、5至3000、5至2000、5至1000、10至10000、10至9000、10至8000、10至7000、10至6000、10至5000、10至4000、10至3000、10至2000、10至1000、100至10000、100至9000、100至8000、100至7000、100至6000、100至5000、100至4000、100至3000、100至2000、100至1000、500至10000、500至9000、500至8000、500至7000、500至6000、500至5000、500至4000、500至3000、500至2000、500至1000、1000至10000、1000至9000、1000至8000、1000至7000、1000至6000、1000至5000、1000至4000、1000至3000、1000至2000个基因组。各可能性代表本发明的单独实施方式。
在一些实施方式中,cfdna的少至0.00001、0.00005、0.0001、0.0005、0.001、0.005、0.01、0.05、0.1、0.2、0.3、0.5、1、2、3、4、5或10%来自所述细胞类型、组织或处于所述细胞状态的细胞。各可能性代表本发明的单独实施方式。在一些实施方式中,cfdna的少至0.1%来自所述细胞类型、组织或处于所述细胞状态的细胞。在一些实施方式中,cfdna的少至1%来自所述细胞类型、组织或处于所述细胞状态的细胞。在一些实施方式中,方法的检测极限是样品中的cfdna的0.1%来自所述细胞类型、所述组织或所述细胞状态。在一些实施方式中,方法的检测极限是样品中的cfdna的1%来自所述细胞类型、所述组织或所述细胞状态。在一些实施方式中,cfdna的少至0.1%来自所述细胞类型、所述组织或所述细胞状态,并且检测到对应于所述细胞类型、组织或细胞状态的至少45个峰。在一些实施方式中,cfdna的少至0.1%来自所述细胞类型、所述组织或所述细胞状态,并且检测到对应于所述细胞类型、组织或细胞状态的至少45-200个峰。在一些实施方式中,cfdna的少至1%来自所述细胞类型、所述组织或所述细胞状态,并且检测到对应于所述细胞类型、组织或细胞状态的至少25个峰。在一些实施方式中,分析至少25个峰提供cfdna的1%来自所述细胞类型、所述组织或所述细胞状态的检测极限。在一些实施方式中,分析至少45个峰提供cfdna的0.1%来自所述细胞类型、所述组织或所述细胞状态的检测极限。在一些实施方式中,分析一定数量(anumberof)的峰包括由至少该数量的峰检测cfdna。在一些实施方式中,cfdna包含0.001-10、0.001-5、0.001-3、0.001-2、0.001-1.5、0.001-1、0.01-10、0.01-5、0.01-3、0.01-2、0.01-1.5、0.01-1、0.1-10、0.1-5、0.1-3、0.1-2、0.1-1.5、0.1-1、0.5-10、0.5-5、0.5-3、0.5-2、0.5-1.50.5-1、1-10、1-5、1-3或1-2%来自所述细胞类型、所述组织或所述细胞状态的cfdna。各可能性代表本发明的单独实施方式。在一些实施方式中,cfdna包含0.1-1%来自所述细胞类型、所述组织或所述细胞状态的cfdna。在一些实施方式中,cfdna包含0.1-3%来自所述细胞类型、所述组织或所述细胞状态的cfdna。
在一些实施方式中,测序是是低深度的。在一些实施方式中,测序的深度小于10亿、7.5亿、5亿、4亿、3亿、2亿、1亿、9000万、8000万、7000万、6000万、5000万、4000万、3000万、2000万、1000万、900万、800万、700万、600万、500万、400万、300万、200万、100万、50万、10万、5万、1万、5000或1000个读段。各可能性代表本发明的单独实施方式。在一些实施方式中,测序的深度小于1000万个读段。在一些实施方式中,测序的深度小于100万个读段。本领域技术人员将理解,随着信息量增加,检测极限下降。此外,随着输入数据量增加(增加试剂(即用于chip的抗体)的数量,增加给定细胞类型/组织/状态的信息性基因座的数量,增加来自所述细胞类型/组织/状态的cfdna的数量),所需的测序深度也因此降低,并且检测极限降低。
在一些实施方式中,所述提供包括提供包含cfdna的体液。在一些实施方式中,所述接触发生在体液中。在一些实施方式中,所述接触包括提供体液和从体液分离cfdna。在一些实施方式中,体液选自:血液、血清、胃液、肠液、唾液、胆汁、肿瘤液、间质液、母乳、脑脊液、尿液、精液、阴道液和粪便。在一些实施方式中,体液是含有cfdna的任何体液。在一些实施方式中,体液是血液。在一些实施方式中,体液是全血、部分溶解的全血、血浆或部分加工的全血中的任一种。
血液样品可以通过标准技术获得,如使用针和注射器。在另一实施方式中,血液样品是外周血样品。可选地,血液样品可以是外周血的分离部分,如血浆样品。在另一实施方式中,在获得血液样品后,可以利用本领域技术人员已知的标准技术从样品提取总dna。在一些实施方式中,在dna提取之前除去完整细胞,从而仅提取自由漂浮的dna。完整细胞可以通过本领域已知的任何方法去除,如作为非限制性实例,通过离心或通过梯度分离,如通过ficol梯度分离。dna提取的非限制性实例是flexigenedna试剂盒(qiagen)。用于接收无细胞dna提取的标准技术是技术人员已知的,其非限制性实例是qiaampcirculatingnucleicacid试剂盒(qiagen)。
在一些实施方式中,测序是下一代测序。下一代测序,也称为高通量测序或大规模并行测序,是实现对来自dna或rna样品的碱基对进行快速高通量测序的任何测序方法。在一些实施方式中,测序是高通量测序。在一些实施方式中,测序是大规模并行测序。这样的测序是本领域公知的,并且可以包括使用illumina阵列、孔和纳米孔测序仪以及离子激流(iontorrent)作为非限制性实例。对于非限制性实例,可以使用诸如illuminanextseq500机器的测序机器,并且可以使用illumina500/550v2试剂盒进行处理。在一些实施方式中,测序是全基因组测序。在一些实施方式中,仅部分基因组被测序。在一些实施方式中,涉及仅部分基因组的芯片或阵列用于下一代测序。
在一些实施方式中,测序是甲基化敏感性测序。在一些实施方式中,本发明的方法在测序之前还包括亚硫酸氢盐转化。在进行测序时,还可以识别dna的甲基化状态,以这种方式,还可以将蛋白质-dna缔合数据与dna甲基化数据组合。这可以提供关于细胞在其死亡时的基因活性的进一步信息,其可以提供对来源细胞或组织或细胞的细胞状态的了解。
在一些实施方式中,本发明的方法可以甚至在来自一种组织/细胞类型的cfdna占全部cfdna的非常小的百分比时用于确定cfdna的来源。在一些实施方式中,某组织和/或细胞类型的cfdna占全部cfdna的少至0.0001%、0.0005%、0.001%、0.005%、0.01%、0.05%、0.1%、0.5%、1%、1.5%、2%、3%、4%、5%、6%、7%、8%、9%或10%。各可能性代表本发明的单独实施方式。在一些实施方式中,某组织和/或细胞类型的cfdna占全部cfdna的多于0.0001%、0.0005%、0.001%、0.005%、0.01%、0.05%、0.1%、0.5%、1%、1.5%、2%、3%、4%、5%、6%、7%、8%、9%或10%。各可能性代表本发明的单独实施方式。在一些实施方式中,某组织和/或细胞类型的cfdna占全部cfdna的少于0.0001%、0.0005%、0.001%、0.005%、0.01%、0.05%、0.1%、0.5%、1%、1.5%、2%、3%、4%、5%、6%、7%、8%、9%或10%。各可能性代表本发明的单独实施方式。在一些实施方式中,某组织和/或细胞类型的cfdna占全部cfdna的0.0001%-10%、0.001%-10%、0.01%-10%、0.1%-10%、0.5%-10%、1%-10%、1.5%-10%、2%-10%、0.0001%-9%、0.001%-9%、0.01%-9%、0.1%-9%、0.5%-9%、1%-9%、1.5%-9%、2%-9%、0.0001%-8%、0.001%-8%、0.01%-8%、0.1%-8%、0.5%-8%、1%-8%、1.5%-8%、2%-8%、0.0001%-7%、0.001%-7%、0.01%-7%、0.1%-7%、0.5%-7%、1%-7%、1.5%-7%、2%-7%、0.0001%-6%、0.001%-6%、0.01%-6%、0.1%-6%、0.5%-6%、1%-6%、1.5%-6%、2%-6%、0.0001%-5%、0.001%-5%、0.01%-5%、0.1%-5%、0.5%-5%、1%-5%、1.5%-5%、2%-5%、0.0001%-4%、0.001%-4%、0.01%-4%、0.1%-4%、0.5%-4%、1%-4%、1.5%-4%、2%-4%、0.0001%-3%、0.001%-3%、0.01%-3%、0.1%-3%、0.5%-3%、1%-3%、1.5%-3%、2%-3%、0.0001%-2%、0.001%-2%、0.01%-2%、0.1%-2%、0.5%-2%、1%-2%、1.5%-2%、0.0001%-1%、0.001%-1%、0.01%-1%、0.1%-1.5%、0.5%-1.5%、0.1%-1%、0.5%-1%、0.0001%-0.1%、0.001%-0.1%或0.0001%-0.001%。各可能性代表本发明的单独实施方式。
在一些实施方式中,所述接触是在包含cfdna的体液中培育试剂。在一些实施方式中,所述接触是在包含cfdna的血液中培育试剂。在一些实施方式中,所述接触是在结合/培育溶液中培育试剂和cfdna。用于进行chip的缓冲剂,具体地培育缓冲剂,是本领域公知的。这种缓冲剂可以从诸如abeam和cellsignalingtechnology的销售chip试剂盒的公司购买。
在一些实施方式中,所述接触伴以恒定的混合进行。在一些实施方式中,所述接触伴以恒定的旋转进行。在一些实施方式中,所述接触在室温或4度下进行。在一些实施方式中,所述接触在冰上进行。在一些实施方式中,所述接触进行至少1、2、3、4、5、6、12、18或24小时。各可能性代表本发明的单独实施方式。在一些实施方式中,所述接触的时间足以使试剂结合至dna缔合蛋白。在一些实施方式中,所述接触的时间足以使试剂结合所提供的dna缔合蛋白的至少10%、20%、30%、40%、50%、60%、70%、80%、90%、95%、97%或99%。各可能性代表本发明的单独实施方式。
在一些实施方式中,本发明的方法用于检测有此需要的对象中的疾病状态或状况,并且其中cfdna来自该对象。在一些实施方式中,本发明的方法用于诊断有此需要的对象中的疾病和/或状况,并且其中cfdna来自该对象。在一些实施方式中,本发明的方法用于诊断疾病或状况的风险增加。本领域技术人员将认识到,很多(若非全部)疾病状态在表现疾病的组织或细胞中诱导细胞死亡。因此,对于细胞死亡来源认识代替该疾病。在一些实施方式中,疾病状态或状况选自心脏疾病或损伤和肝脏疾病或损伤。在一些实施方式中,疾病状态或状况选自心脏疾病或损伤、肝脏疾病或损伤和癌症。在一些实施方式中,疾病状态是癌症。在一些实施方式中,疾病状态是癌前状态。在一些实施方式中,疾病状态是癌症或癌前状态。在一些实施方式中,疾病状态或状况选自心跳停止和肝休克。在一些实施方式中,疾病状态是脑损伤。在一些实施方式中,疾病状态是菌血症。在一些实施方式中,疾病状态是感染。在一些实施方式中,疾病状态或状况选自癌症、神经变性疾病、感染、组织损伤、炎症、自身免疫疾病、关节炎、肝脏炎症、肠炎症、自身免疫疾病、菌血症、药物副作用引起的组织损伤、组织坏死、和糖尿病。在一些实施方式中,神经变性疾病是帕金森氏病或阿尔茨海默氏病。在一些实施方式中,自身免疫疾病是狼疮或多发性硬化症。在一些实施方式中,疾病是癌症,并且本发明的方法确定癌症的来源细胞或组织。本领域技术人员将充分理解,指示来自癌基因的cfdna的活跃转录和序列的蛋白质的缔合指示癌性或癌前状态。此外,指示来自肿瘤抑制剂的cfdna的转录沉默和序列的蛋白质的缔合还指示癌性或癌前状态。类似地,癌基因和肿瘤抑制剂的增强子区域的激活或阻遏分别也指示癌性或癌前状态。
在一些实施方式中,本发明的方法进一步包括使用与第二dna缔合蛋白结合的试剂再次执行步骤a-d,并且其中第二dna缔合蛋白是与已使用的dna缔合蛋白不同的蛋白质。在一些实施方式中,第二dna缔合蛋白不同于第一dna缔合蛋白。在一些实施方式中,本发明的方法可以重复至少2、3、4、5、6、7、8、9或10次,其中每次结合不同的dna缔合蛋白。各可能性代表本发明的单独实施方式。
在一些实施方式中,本发明的方法包括使样品与至少2种试剂接触,其中各试剂结合至物理载体,并且载体包含对各试剂独特的短dna标签,其中在对分离的cfdna进行测序后,短dna标签识别使cfdna分离的试剂。在一些实施方式中,在测序之前将dna标签连接至cfdna分子。在一些实施方式中,载体是缀合至单一试剂和对试剂独特的短dna标签的珠。在一些实施方式中,试剂是抗体,并且短dna标签是dna条形码。这可以例如是共价结合chip抗体(如h3k4me1)并结合用于识别h3k4me1缔合的dna的条形码的顺磁珠,以及同时被加入样品中、同时与cfdna连接和同时测序的共价结合chip抗体(如h3k4me3)并结合识别h3k4me3缔合的dna的条形码的其他顺磁珠。
在一些实施方式中,方法进一步包括治疗对象。在一些实施方式中,治疗针对被检测到的疾病。在一些实施方式中,治疗是基于在对象中的死亡细胞的细胞状态、来源组织、细胞类型或其组合的适当治疗。本领域技术人员将理解,如果例如在具体器官中发现癌症,则可以针对该类型的癌症调整治疗。类似地,如果某具体途径在癌症或疾病中活跃,则一种治疗方式可能比另一种更适合。例如,长的非编码rnaegfr-as1(其介导癌症对egfr的成瘾(canceraddictiontoegfr),并且在被高度表达时可以通过抗egfr抗体治疗使肿瘤对egfr抑制不敏感)的活跃转录的检出将表明应避免egfr抑制性治疗。
疾病检测
另一方面,提供了检测对象的疾病状态的方法,该方法包括:
a.提供来自对象的样品,其中所述样品包含cfdna;
b.使样品与结合dna缔合蛋白的至少一种试剂接触;
c.分离该试剂及其任何结合的蛋白质和cfdna;
d.对分离的cfdna进行测序;和
e.将包含疾病相关突变的cfdna分子指定为源自处于疾病状态的细胞;
从而检测对象的疾病状态。
另一方面,提供了用于改进在来自对象的cfdna中的疾病检测的方法,该方法包括在对来自对象的cfdna进行染色质免疫沉淀,然后在免疫沉淀的cfdna中进行疾病检测。
在一些实施方式中,染色质免疫沉淀包括:
a.使来自对象的cfdna与结合dna缔合蛋白的至少一种试剂接触;和
b.分离该试剂及其任何结合的蛋白质和cfdna。
在一些实施方式中,疾病检测包括cfdna的测序。在一些实施方式中,疾病检测包括来自对象的cfdna的测序。在一些实施方式中,疾病检测包括免疫沉淀的cfdna的测序。在一些实施方式中,疾病检测和/或测序包括cfdna的扩增。在一些实施方式中,疾病检测和/或测序不包括cfdna的扩增。在一些实施方式中,来自对象的cfdna中的疾病检测包括cfdna的扩增。在一些实施方式中,非改进的疾病检测包括cfdna的扩增。在一些实施方式中,免疫沉淀的cfdna中的疾病检测不包括免疫沉淀的cfdna的扩增。在一些实施方式中,改进的疾病检测不包括免疫沉淀的cfdna的扩增。在一些实施方式中,扩增是pcr扩增。在一些实施方式中,扩增是非特异性扩增。在一些实施方式中,扩增是疾病相关序列的扩增。
如本文所用,“疾病相关突变”是指已知引起或增加罹患疾病的风险的dna突变。疾病相关突变是公知的,包括例如,囊性纤维化中的cftr的1522a、1523t和1524c的缺失(f508缺失);gaucher病中的gba基因座的1226a至g(n370s);α1-抗胰蛋白酶缺乏症中的serpina1的突变;β地中海贫血中的hbb的突变;以及阿尔茨海默氏病中的psen1的突变。在癌症中已知多种疾病相关突变,一些是多种癌症类型共有的,一些是特定癌症特有的。p53、myc、bref、brca(仅举几例)中的突变是本领域公知的。还可以研究成组的疾病相关突变。在一些实施方式中,研究至少1、2、3、5、7、10、12、15、17或10种突变。各可能性代表本发明的单独实施方式。在一些实施方式中,代替测序,采用以突变特异性引物进行的pcr。可以采用任何检测dna突变的方法来代替测序;但是,测序具有同时检查多种突变(包括一组突变)的优点。
在一些实施方式中,dna缔合蛋白与疾病相关突变的缔合指示疾病状态。本领域技术人员将充分理解,指示基因编码区域中的活跃转录突变的蛋白质的缔合将指示突变基因正在被转录并且将指示癌性或癌前状态。类似地,调控区域中的突变将与指示该调控区域的蛋白质缔合,因此也将指示癌性或癌前状态。
在一些实施方式中,疾病相关突变处于基因的编码区域中,并且dna缔合蛋白与dna的缔合指示活跃转录。在一些实施方式中,该基因是癌基因或肿瘤抑制基因。在一些实施方式中,疾病相关突变处于调控区域中,并且dna缔合蛋白与dna的缔合指示调控区域。
在一些实施方式中,本发明的方法进一步包括利用与第二dna缔合蛋白结合的试剂再次进行步骤b-e,并且其中第二dna缔合蛋白不同于第一dna缔合蛋白。如果将要研究多种突变并且其位于不同的基因组区域(例如基因体和增强子),则可以针对不同的dna缔合蛋白重复进行chip。
仅一部分cfdna的免疫沉淀(如通过h3k36me3免疫沉淀富集活跃转录基因的基因体序列)大大提高了信息性dna的浓度。由此,测序可以显著更便宜地并且使用更少的试剂进行。此外,该方法允许检测与特定基因组注释(annotations)(活性基因、活性启动子、活性增强子等)相关的突变,而无需预先限定有限的一组基因组位置(如一组癌症风险基因)和设计特定试剂以扩增和/或检测那些预先限定的序列。通过首先减少基因组测序部分的有效尺寸(减少至仅免疫沉淀部分),大大降低了测序成本。此外,由于重复序列和非信息性序列较少,因此背景较少并且假阳性结果较少。最后,给定深度的测序提供了多于信息性序列的读段。
在一些实施方式中,所述改进包括以下至少一项:降低信噪比,增加对阳性疾病检出的置信度,减少对伪疾病检出,以及用较少的来自对象的cfdna准确地检测疾病。在一些实施方式中,所述增加是至少10、20、30、40、50、60、70、80、90、100、200、300、400、500或1000%增加。各可能性代表本发明的单独实施方式。在一些实施方式中,该减少是至少10、20、30、40、50、60、70、80、90、85、97、99或100%减少。各可能性代表本发明的单独实施方式。
在一些实施方式中,所述较少的来自对象的cfdna少于1000、900、800、700、600、500、400、300、200、100、90、80、70、60或50ng的cfdna。各可能性代表本发明的单独实施方式。在一些实施方式中,可以用与较大量的cfdna相比较少的cfdna实现相同的准确性。在一些实施方式中,与用较大量的cfdna实现的覆盖范围相比,突变处相同的测序覆盖范围可用较少的cfdna实现。
计算机程序产品
另一方面,提供了用于确定无细胞dna(cfdna)的来源细胞或组织的计算机程序产品,包括其上包含程序代码的非暂时性计算机可读存储介质,该程序代码可被至少一个硬件处理器,从而:
a.测序或访问用结合dna缔合蛋白的试剂分离的cfdna的测序;
b.将来自该cfdna的cfdna分子分配给某来源细胞或组织——通过将该分子的dna序列与所述细胞类型或组织中与dna缔合蛋白缔合的序列进行比较;和
c.提供关于cfdna的来源细胞或组织的输出。
另一方面,提供了用于确定对象中的细胞在该细胞死亡时的细胞状态的计算机程序产品,包括其上包含程序代码的非暂时性计算机可读存储介质,该程序代码可被至少一个硬件处理器执行,从而:
a.测序或访问用结合dna缔合蛋白的试剂分离的来自对象的cfdna的测序;
b.将来自该cfdna的cfdna分子分配给某细胞状态——通过将该分子的dna序列与所述细胞状态中与dna缔合蛋白缔合的序列进行比较;和
c.提供关于对象中的细胞在该细胞死亡时的细胞状态的输出。
另一方面,提供了用于确定cfdna的来源细胞或组织的系统,包括:
a.用于测序用结合dna缔合蛋白的试剂分离的cfdna的一个或多个装置;
b.处理器;和
c.存储介质,其包括计算机应用程序,该计算机应用程序在被处理器执行时被配置以:
i.测序或访问用结合dna缔合蛋白的试剂分离的cfdna的测序;
ii.将来自该cfdna的cfdna分子分配给某来源细胞或组织——通过将该分子的dna序列与所述细胞类型或组织中与dna缔合蛋白缔合的序列进行比较;和
iii.从处理器输出cfdna的来源细胞或组织。
另一方面,提供了用于确定对象中的细胞在该细胞死亡时的细胞状态的系统,包括:
a.用于测序用结合dna缔合蛋白的试剂分离的cfdna的一个或多个装置;
b.处理器;和
c.存储介质,其包括计算机应用程序,该计算机应用程序在被处理器执行时被配置以:
i.测序或访问用结合dna缔合蛋白的试剂分离的cfdna的测序;
ii.将来自该cfdna的cfdna分子分配给某细胞状态——通过将该分子的dna序列与所述细胞状态中与dna缔合蛋白缔合的序列进行比较;和
iii.从处理器输出cfdna的来源细胞或组织。
另一方面,提供了用于检测对象的疾病状态的计算机程序产品,包括其上包含程序代码的非暂时性计算机可读存储介质,该程序代码可被至少一个硬件处理器执行,从而
a.将来自该cfdna的cfdna分子分配给某疾病状态——通过将该分子的dna序列与和该疾病状态相关的突变序列进行比较;
b.提供关于对象的疾病状态的输出。
计算机可读存储介质可以是有形装置,其可以保留和存储供指令执行装置使用的指令。计算机可读存储介质可以是例如但不限于电子存储装置、磁存储装置、光存储装置、电磁存储装置、半导体存储装置、或前述的任何适当组合。计算机可读存储介质的更具体实例的非穷举列举包括以下:便携式计算机磁盘(软盘,diskette)、硬盘、随机存取存储器(ram)、只读存储器(rom)、可擦可编程只读存储器(eprom或闪存)、静态随机存取存储器(sram)、便携式压缩盘只读存储器(cd-rom)、数字多功能磁盘(dvd)、记忆棒、软盘(afloppydisk)、机械编码装置,如打孔卡或其上记录指令的凹槽中凸起结构、以及前述的任何适当组合。如本文所用,计算机可读存储介质将不被理解为本身是暂时性号,如无线电波或其他自由传播的电磁波、通过波导或其他传输介质传播的电磁波(例如,穿过光纤电缆的光脉冲)或通过电线传输的电信号。
本文描述的计算机可读程序指令可以从计算机可读存储介质被下载到相应的计算/处理装置,或经由例如因特网、局域网、广域网和/或无线网络的网络被下载到外部计算机或外部存储装置。网络可以包括铜传输电缆、光传输光纤、无线传输、路由器、防火墙、交换机、网关计算机和/或边缘服务器。各计算/处理装置中的网络适配器卡或网络接口从网络接收计算机可读程序指令,并发送计算机可读程序指令以存储在相应计算/处理装置内的计算机可读存储介质中。
用于执行本发明的操作的计算机可读程序指令可以是汇编指令、指令集架构(instruction-set-architecture)(isa)指令、机器指令、机器依赖性指令、微代码、固件指令、状态设置数据或用一种或多种编程语言(包括目标定向编程语言、如java、smalltalk、c++等)和常规程序编程语言(如“c”编程语言或类似编程语言)的任意组合编写的源代码或目标代码。计算机可读程序指令可以完全在用户计算机上执行,部分在用户计算机上执行,作为独立软件包执行,部分在用户计算机上并且部分在远程计算机上执行,或完全在远程计算机或服务器上执行。在后者情况下,远程计算机可以通过任何类型的网络(包括局域网(lan)或广域网(wan))连接到用户计算机,或者可以与外部计算机建立连接(例如,通过internet,利用internet服务提供商)。在一些实施方式中,电子电路——包括例如可编程逻辑电路,现场可编程门阵列(fpga)或可编程逻辑阵列(pla)——可以通过利用计算机可读程序指令的状态信息而个性化电子电路来执行计算机可读程序指令,以执行本发明的方方面面。
可以将这些计算机可读程序指令提供给通用计算机、专用计算机或其他可编程数据处理设备的处理器,以产生机器,使得该指令(其通过计算机或其他可编程数据处理设备的处理器执行)建立用于实现流程图和/或框图一个或多个框中指定的功能/行动的手段。这些计算机可读程序指令还可以被存储在这样的计算机可读存储介质中:其可以指导计算机、可编程数据处理设备和/或其他装置以具体方式工作,使得其中存储了指令的计算机可读存储介质包括包含实现流程图和/或框图一个或多个框中指定的功能/行动的方方面面的指令的制品。
实施方式可以包括实现本文描述和示例的功能的计算机程序,其中计算机程序在包括存储在机器可读介质中的指令和执行该指令的处理器的计算机系统中实施。然而,显然,在计算机编程中可以有多种不同的方式实现实施方式,并且实施方式不应被解释为限于任一组计算机程序指令。此外,技术程序员将能够编写实现本文描述的一个或多个公开实施方式的计算机程序。因此,认为具体一组程序代码指令的公开对于充分理解如何建立和使用实施方式而言是不必要的。此外,本领域技术人员将理解,本文描述的实施方式的一个或多个方面可以通过硬件、软件或其组合来执行,如可以在一个或多个计算系统中实施。而且,对由计算机执行的行动的任何提及用都不应被解释为由单一计算机执行,因为多于一个计算机可以执行该行动。
用于测序的装置是指允许一段dna的序列被确定的部件组合。在一些实施方式中,测试装置允许dna的高通量测序。在一些实施方式中,测试装置允许dna的大规模并行测序。所述部件可包括以上关于测序方法所述的那些中的任意者。
在某些实施方式中,系统或测试套件还包括用于处理器输出的显示器。
多重化
另一方面,提供了固体载体,其包含捕获剂和条形码化试剂。
如本文所用,术语“捕获剂”是指与蛋白质结合并因此可以将蛋白质捕获并保留至固体载体的分子。在一些实施方式中,捕获剂是小分子。在一些实施方式中,捕获剂是蛋白质。在一些实施方式中,捕获蛋白通过蛋白-蛋白相互作用捕获第二蛋白。在一些实施方式中,捕获蛋白是抗体或其抗原结合片段。捕获剂可以是特异性结合染色质或核酸的任何分子。
如本文所用,术语“条形码化试剂”是指包含独特分子或部分的任何底物,所述独特分子或部分可以用作识别目标分子的条形码。条形码是本领域公知的,并且任何足够独特以识别目标分子的分子或部分都可以用作条形码。在一些实施方式中,条形码化试剂是条形码本身。在一些实施方式中,条形码是蛋白质条形码。在一些实施方式中,条形码是蛋白质标签。在一些实施方式中,条形码是荧光蛋白。
在一些实施方式中,条形码是核酸条形码。在一些实施方式中,核酸分子是短核酸分子。在一些实施方式中,其长度少于3、5、7、10、12、15、17、20或25个核苷酸。各可能性代表本发明的单独实施方式。在一些实施方式中,核酸分子长度为3至10、3至15、3至20、3至25、3至30、3至35、3至40、4至45、3至50、5至10、5至15、5至20、5至25、5至30、5至35、5至40、5至45或5至50个核苷酸。各可能性代表本发明的单独实施方式。在一些实施方式中,条形码化试剂是用于将条形码连接至目标分子的酶。在一些实施方式中,条形码化试剂是连接酶。在一些实施方式中,条形码化试剂是条形码,并且固相载体还包含用于将条形码连接至目标分子的酶。
在一些实施方式中,目标分子是蛋白质。在一些实施方式中,目标分子是核酸分子。在一些实施方式中,dna或rna中的目标分子。在一些实施方式中,捕获剂捕获与目标分子缔合的蛋白质。在一些实施方式中,捕获剂捕获dna缔合蛋白,并且目标分子是dna。在一些实施方式中,dna是cfdna。在一些实施方式中,目标分子与捕获剂捕获的蛋白质复合。
固体载体可以是可附着生物大分子的任何聚合物或无机材料。附着可以是直接或间接的。在一些实施方式中,固体载体由用于组装微流体装置的材料制成。在一些实施方式中,固体载体是珠。在一些实施方式中,珠是琼脂糖珠。在一些实施方式中,固体载体是磁珠或顺磁珠。在一些实施方式中,固体载体是琼脂糖珠或磁珠或顺磁珠。在一些实施方式中,载体与捕获剂缀合。在一些实施方式中,载体与chip抗体缀合。在一些实施方式中,载体与条形码化试剂缀合。在一些实施方式中,载体与捕获剂和条形码化试剂缀合。缀合可以通过本领域已知的任何方法进行,包括但不限于共价键合、基于电荷的键合和疏水相互作用。在一些实施方式中,缀合是生物素与抗生物素蛋白(avidin)的缀合。在一些实施方式中,缀合通过胺结合技术进行。在一些实施方式中,胺结合技术是环氧。在一些实施方式中,缀合是通过羧基捕获。
另一方面,提供了对单一溶液中的多于一种目标分子多重化测定的方法,该方法包括:
a.在溶液中将第一目标分子捕获至本发明的第一固体载体;
b.在所述溶液中将至少第二目标分子捕获至本发明的第二固体载体;
c.将第一目标分子和第一条形码以及至少第二目标分子和第二条形码附着;
d.同时对第一和第二目标分子进行测定,其中第一目标分子的测定结果通过第一条形码识别,并且第二目标分子的测定结果通过第二条形码识别;
从而对单一溶液中的多于一种目标分子进行多重化测定。
如本文所用,“多重化测定”是指同时对多个样品进行一种测定。多重化在样品有限时是有用的,因为该测定在时间、金钱、试剂或样品输入方面是昂贵的。通过利用本发明的方法进行多重化,可以从始至终同时进行测定,从而减少样品之间的差异(图5b)。例如,在多重化染色质免疫沉淀然后利用本发明的方法进行下一代测序(chip-seq)测定时,在一个管中一次进行全部所用抗体的蛋白质捕获。条形码的连接也全部一次发生,并且洗涤和测序也都一体进行。这极大地限制了测定性能的任何样品间差异。在一些实施方式中,测定是chip、chip-seq、cfchip、cfchip-seq、蛋白质定量和蛋白质-蛋白质相互作用测定中的任一种。蛋白质定量可通过向蛋白质添加通用dna接头/序列,然后连接条形码来实现。在一些实施方式中,本发明的方法进一步包括将接头附接至目标蛋白质。在一些实施方式中,测定是染色质免疫沉淀然后测序(chip-seq)。
在一些实施方式中,目标分子是蛋白质。在一些实施方式中,目标分子是核酸分子。在一些实施方式中,目标分子是蛋白质和/或核酸分子。
在一些实施方式中,通过条形码识别包括对目标分子的数量和/或数目的定量。在一些实施方式中,条形码的数量和/或数目等于目标分子的数量和/或数目。在一些实施方式中,条形码的数量和/或数目与目标分子的数量和/或数目成比例。在一些实施方式中,条形码的数量和/或数目与目标分子的数量和/或数目相等或成比例。
如本文所用,术语“约”当与值结合时是指参考值±10%。例如,约1000纳米(nm)的长度是指1000nm±100nm的长度。
注意,如本文和所附权利要求书中所用,单数形式“一个(a)”、“一种(an)”和“所述(the)”包括复数指代,除非上下文另有明确指出。因此,例如,“一个多核苷酸”的提及包括多个这样的多核苷酸,并且对“所述多肽”的提及包括对一种或多种多肽及本领域技术人员已知的其等同形式的提及,等等。还应注意,权利要求可能被撰写为不包括任何可选的要素。因此,这种陈述与权利要求要素的记载或“消极”限制的使用结合旨在作为使用诸如“仅”、“只有”等排他性术语的先行基础。
在那些使用类似于“a、b和c中的至少一个等”的常用语的情况下,总体上这样的结构意图是本领域技术人员可以理解该常用语的意义(例如,“具有a、b和c中至少一个的系统”包括但不限于具有单独a,具有单独b,具有单独c,具有a和b,具有a和c,具有b和c,和/或具有a、b和c的系统等)。本领域技术人员将进一步理解,实际上,无论是在说明书、权利要求书还是附图中,呈现两个或更多个替代性术语的任何析取词语和/或短语都应被理解为考虑包括多个术语中的一个、两个术语中的一个或两个术语中的两个的可能性。例如,短语“a或b”将被理解为包括“a”或“b”或“a和b”的可能性。
应当理解,为清楚起见在分开的实施方式的情况下描述的本发明的某些特征也可以在单一实施方式中组合提供。相反,为简洁起见在单一实施方式的情况下描述的本发明的各种特征也可以分开地或以任何合适的子组合来提供。与本发明有关的实施方式的所有组合都被本发明具体地涵盖,并且在本文中被公开如同各个和每一个组合都被单独地并且明确地公开。另外,各种实施方式及其要素的所有子组合也被本发明具体地涵盖,并且在本文中被公开如同各个和每一个这样的子组合在本文中都被分开地并且明确地公开。
在考察以下实施例之后,本发明的其他目的、优点和新颖特征对于本领域普通技术人员将显而易见,该实施例并非意图是限制性的。另外,如上文所述且如所附权利要求书部分所述,本发明的各种实施方式和方面中的每一个均在以下实施例中得到实验支持。
上文描述的以及如所附下面权利要求书部分所述的本发明的各种实施方式和方面在以下实施例中得到实验支持。
实施例
通常,本文所用的命名法以及本发明中使用的实验室程序包括分子、生物化学、微生物学和重组dna技术。这种技术在文献中有详尽的解释。参见,例如,"molecularcloning:alaboratorymanual"sambrooketal.,(1989);"currentprotocolsinmolecularbiology"volumesi-iiiausubel,r.m.,ed.(1994);ausubeletal.,"currentprotocolsinmolecularbiology",johnwileyandsons,baltimore,maryland(1989);perbal,"apracticalguidetomolecularcloning",johnwiley&sons,newyork(1988);watsonetal.,"recombinantdna",scientificamericanbooks,newyork;birrenetal.(eds)"genomeanalysis:alaboratorymanualseries",vols.1-4,coldspringharborlaboratorypress,newyork(1998);美国专利号4,666,828;4,683,202;4,801,531;5,192,659和5,272,057中提出的方法;"cellbiology:alaboratoryhandbook",volumesi-iiicellis,j.e.,ed.(1994);"cultureofanimalcells-amanualofbasictechnique"byfreshney,wiley-liss,n.y.(1994),thirdedition;"currentprotocolsinimmunology"volumesi-iiicoliganj.e.,ed.(1994);stitesetal.(eds),"basicandclinicalimmunology"(8thedition),appleton&lange,norwalk,ct(1994);mishellandshiigi(eds),"strategiesforproteinpurificationandcharacterization-alaboratorycoursemanual"cshlpress(1996);其全部通过引用并入。贯穿本文提供了其他一般参考文献。
材料和方法
患者
所有临床研究均获得了当地有关伦理委员会的批准。该研究得到了hebrewuniversity-hadassahmedicalcenterofjerusalem的伦理委员会的批准。在采血之前,从所有对象或其法定监护人获得了知情同意。
样品采集
将血样收集在
珠制备
按照制造商的说明,将50μg抗体与5mg环氧m270dynabeads(invitrogen)缀合。将抗体-珠复合物于4℃保存在pbs,0.02%叠氮化物溶液中。
免疫沉淀、ngs文库制备和测序
每个cfchip样品使用0.2mg缀合珠(~2μg抗体)。将抗体-珠复合物直接加入血浆(1-2ml血浆),并通过在4℃旋转过夜使其与cf-核小体结合。将珠磁化,并用血液洗涤缓冲剂(bwb50mmtris-hcl,150mmnacl,1%tritonx-100、0.1%脱氧胆酸钠,2mmedta,1×蛋白酶抑制剂混合物)洗涤6次,用bwb-500(与仅具有500mmnacl的bwb相同)洗涤两次,并且使用10mmtrisph7.4洗涤3次。在冰上用150ul缓冲剂通过在磁体上使珠侧至侧(sidetoside)移动而进行所有洗涤。在不含清洁剂的缓冲剂中洗涤的过程中,不利用真空除去上清液。除去珠后,将血浆储存起来,因为其适于进行更多轮的cfchip。
进行珠上染色质条形码化和文库扩增以克服低输入材料的问题。此程序支持由少至1000个细胞准备cfchip。以下步骤全部对珠执行,以减少cfdna释放和试管转换过程中可能发生的cfdna损失。通过t4dna聚合酶和t4多核苷酸激酶修复dna末端。洗涤后,利用klenowexominus将腺嘌呤碱基添加到dna的修复末端。再次洗涤后,将dna接头连接在上;在这种情况下,使用具有dna条形码序列的illumina接头。对于dna洗脱和净化步骤,将珠在55℃下在补充有50单位的蛋白酶k(epicenter)的50μl染色质洗脱缓冲剂(10mmtrisph8.0,5mmedta,300mmnacl,0.6%sds)中温育1小时,并通过1.2xspri净化(ampurexp,agencourt)纯化dna。将纯化的dna在25μleb(10mmtrisph8.0)中洗脱,并将23μl洗脱的dna用于用kapa热启动聚合酶进行的pcr扩增(16个循环)。通过1.2xspri净化纯化扩增的dna,并在12μleb中洗脱。通过qubit测量洗脱的dna浓度,并通过tapestation可视化分析片段大小。注意:如果在文库扩增后接头二聚体仍是通过tapestation可见的,则可以将条形码不同的样品合并在一起,在4%琼脂糖凝胶(
序列分析
使用带有“no-mixed”和“”特征(标示,flag)的bowtie2将读段与人基因组(hg19)进行比对。我们舍弃了比对得分低和片段重复的读段。
roadmapepigenome图谱
我们从roadmapepigenomeconsortium数据库(egg2.wustl.edu/roadmap/data/byfiletype/alignments/consolidated/)下载了整合的比对数据。在这些中,我们添加了肾脏样品(egg2.wustl.edu/roadmap/data/byfiletype/alignments/unconsolidated/h3k4me3/bi.adult_kidney.h3k4me3.27.filt.tagalign.gz和egg2.wustl.edu/roadmap/data/byfiletype/alignments/unconsolidated/h3k4me3/bi.adult_kidney.h3k4me3.153.filt.tagalign.gz)。对于我们的分析,我们舍弃了产前、esc和细胞系样品,结果得到了71种组织和细胞类型。
肿瘤型基因特征
我们从xena项目分析的tcga和gtex项目下载了rna-seq数据(toil实现了可重现的、开放来源的大型生物医学数据分析,vivianj,etal.,nat.biotechnol.,2017,以及toilrnaseqrecompute数据库,tcga-data.nci.nih.gov)。我们定义了在一种肿瘤类型中过表达的基因集满足三个要求:1)与相应组织样品相比,肿瘤样品中的表达显著更高(t检验,fdr校正后q<0.001);2)与所有健康样品相比,其表达显著更高(t检验,fdr校正后q<0.001);3)肿瘤中的中值表达高于各健康样品中的中值表达。
tss位置目录
我们下载了所有整合组织的roadmapepigenomeconsortiumchromhmmchromhmm注释(egg2.wustl.edu/roadmap/data/byfiletype/chromhmmsegmentations/chmmmodels/coremarks/jointmodel/final/all.mnemonics.bedfiles.tgz)。利用这些注释,我们构建了潜在tss位点的目录。我们将该目录扩展到包括ucsc基因数据库和ensembf转录体数据库中的注释转录体的tss为中心的3kb区域(ucsc已知基因:bioconductorannotationhubah5036;ensembl转录体bioconductorannotationhubah5046;基因组注释:bioconductorannotationhubah5040)。我们使用组合的目录来限定沿基因组的区域,这些区域是tss或“背景”(最可能不是tss)。后面的区域通过5kb尺寸的窗口分片化(瓦片化,tiled)。
我们定量了目录中各样品和图谱样品中覆盖各区域的读段数目。我们估测了各样品沿基因组的非特异性读段的局部适应性模型,并提取了代表各样品的目录中的特定chip信号的计数(参见下文)。然后将这些样品标准化(补充文本),并在参考健康样品中缩放至(scaledto)1m读段。
组织/过程特征
为了限定特定修饰的组织特异性特征,我们考察了图谱的bin化(binned)表示。对于各组织,我们限定了具有在目标组织的样品中的一个中的信号而不覆盖所有其他样品的独特窗口的特征(参见下文)。
为了限定过程特征,我们通过在注释中包括与基因启动子重叠的所有窗口,将基因特异性注释(例如go)转换成基因组窗口。
统计学分析
我们考虑两种不同的统计学检验。对于这两个测试,我们需要估测背景覆盖,即来自非特异性下拉的读段(参见下文)。
第一测试是特征是否存在。形式上,我们考察了是否可以否定零假设,即特征窗口中的读段数目将是根据背景率的泊松分布(参见下文)。我们计算特征窗口中观察到的读段的实际数目的p值,为根据零假设具有该数目或更高数目的概率。对于特异性特征的零假设的否定表明,特征中的一些窗口携带构成cf-核小体库(pool)的细胞亚群中的在论(inquestion)修饰。
第二测试是特征是否与在健康基线对象中所预期的相比过表达。为了限定后者预期(值),我们使用来自5个健康样品的平均信号来限定各窗口中的平均读段数(每百万)。我们然后估测两个样品特异性参数——第一是背景率(如上讨论),第二是对平均预期值重新缩放(rescale)至特定样品的测序深度的比例因数(换算系数,scalingfactor)(参见下文)。这些共同限定了在零假设(即对象来自健康人群)下各窗口中的预期覆盖。我们计算特征窗口中的观察到的读段的实际数目的p值,为根据零假设具有该数目或更高数目的概率。对于特异性特征的零假设的否定表明,特征中的一些窗口具有比我们预期健康对象中的信号更高的信号。解释是,这些是构成对象cf-核小体库的细胞中的活跃异常过程。
tss位置目录
我们通过以下步骤构建了tss目录。所有步骤均基于人基因组版本“hg19”进行:
1.我们从roadmapepigenomics网站下载了整个人类基因组中111种组织和细胞类型的chromhmm调用(calls)(egg2.wustl.edu/roadmap/data/bydatatype/rna/expression/57epigenomes.rpkm.pc.gz)。下载了ucsc浏览器已知的基因注释和ensembl转录体注释(ucsc已知基因:bioconductorannotationhubah5036;ensembl转录体bioconductorannotationhubah5046;基因组注释:bioconductorannotationhubah5040)。
2.我们过滤了所有标有状态“l_tssa”或“2_tssaflnk”的基因组范围,并合并了在完全同组的组织中标为任一状态的相邻范围。我们称这些为“chromhmmtss窗口”。我们找到了476,931个这样的窗口。通过以下步骤,对各chromhmmtss窗口分配基因名称(一个或多个)。
a.如果其位于ucsc已知基因注释中的一个或多个tss的2.5kb之内,则其被分配这些基因的名称。
b.如果不是这样,我们搜索了2.5kb以内的ensembl转录体起点。再次,如果发现这样,则tss窗口接收与转录体相关的基因名称。
c.所有其他tss窗口仍无命名。
3.为了包括tss目录中未显示的转录体,我们考察了ucsc已知基因数据库中的所有基因和ensembl数据库中的所有转录体。对于各者,我们都定义了以tss为中心的3kb尺寸的tss窗口。我们舍弃了所有与步骤2中的tss窗口重叠的这种窗口。此步骤分别从ucsc已知基因和ensembl转录体分别添加了总共14,857和41,376个tss窗口。
4.我们创建了在tss窗口之间分片化其余基因组区域的窗口。对于没有相邻tss窗口的各tss窗口,我们创建了1kb(或更小)尺寸的“侧接”区域。这产生370,332个侧接窗口(因为根据不同组织中的chromhmm调用,一些tss窗口彼此相邻)。其余未覆盖区域以5kb(或更小)尺寸的“背景”区域分片化。总共有502,263个这种窗口。
所得目录被另存为bed文件(tss.bed)。
测序文件的处理
使用bcl2fastq(2.18)进行碱基调用。使用带有“no-mixed”和“no-discordant”特征的bowtie2将配对末端的读段映射到人类基因组(hg19),舍弃质量为0的读段。使用带有“bedpe”特征的bedtools“bamtobed”获得bedpe文件(各片段的起点和终点),舍弃重复的片段。使用bedtools“intersect”命令以及使用bioconductor“genomicranges”countoverlaps()函数,将bedpe文件转换为目录中窗口的覆盖计数。两种方法都为各窗口计数与窗口重叠的测序片段的数量。
估测背景信号
各chip程序具有非特异性背景信号。在cfchip情况下,背景是由于dna和染色质片段与珠-抗体复合物的一些形式的非特异性结合所致。我们的经验表明,在样品和珠-抗体连接批次之间背景水平不同。此外,样品之间测序深度不同,并且在深度测序的样品中,背景读段的数量增加。因此,估测背景信号水平以能够使其与实际信号形成对比是重要的。
最初,我们采用了一种简单的程序以去除h3k4me3信号中的背景。我们认为,h3k4me3中几乎所有的特异性信号都在tss和基因5'区域处。因此,其他位置的读段代表背景。为了说明在我们的tss目录中未注释的tss,我们认为背景窗口的某小部分可能包含真实信号,因此我们除去了具有最高值的那些。
更具体地,我们进行了以下内容。我们创建了一个矢量,其中覆盖了所有尺寸≥4kb的“背景”窗口(549,385个中的421,465个)。并应用了以下程序:
此程序对于除去离群窗口的分位数选择相对可靠。
但是,在某些样品中,泊松分布不是背景值的良好适配。进一步考察表明,这种不一致在很大程度上是由于局部背景影响所致。一个局部影响是在男性中以50%的水平而在女性中以100%(x)和0%(y)出现出现的性染色体。这些不是唯一的局部影响——一些区域显示出较高水平的背景。这可能是由于分段重复(靠近着丝粒和端粒的区域)或可及性问题引起的。此外,在癌症样品中,存在明显的患者特异性偏差。
为了克服这些问题,我们设计了局部背景率估测值。我们使用了上述估测程序,但以连续水平的分辨率。
1.基因组范围的背景水平。
2.染色体特异性背景。
3.以2.5mb的偏移量覆盖各染色体的10mb分片(tiles)。
4.以l.25mb的偏移量覆盖各染色体的5mb分片。
各水平的估测使用前一水平的估测值作为先验(在水平2和3中使用1000个窗口的伪计数,在水平4中使用500个窗口的伪计数)。
结果是在5mb重叠分片下的背景覆盖率估测值。为了获得单个估测值,对于各位置,我们采用覆盖其的分片(一般4个分片)的估测值的最大值。我们选择最大值是因为我们认为背景的过高估测可能减少估测的信号,但会减少背景伪像的数量。图6a显示了健康男性样品的背景估测。图6b显示了健康女性样品,其中chrx背景低于常染色体(略大于一半),而chry背景更低一点。chry中的许多位置与chrx中的位置直系同源,导致估测值偏离。其他偏差发生在着丝粒附近,我们发现在此一些染色体(例如chrl、chr9)中的背景水平较高。对癌症患者进行考察时,背景估测值的可变性要大得多,大概反映了肿瘤中的染色体异常(图6c)。
基因水平信号和标准化
对于各基因,我们分配了一组用基因名称注释的tss窗口。对于各样品,我们计算了分配给该基因的窗口的实际总覆盖率以及这些窗口的背景读段的预期平均值(使用各窗口处可能不同的局部比率和窗口尺寸)。
更确切地,
其中wg是分配给基因g的窗口集合,c[w,s]是样品s中窗口w的覆盖率,并且λ^[w,s]是样品s中窗口w的估测背景率。
零假设是覆盖率cg以参数gb按照泊松分布。因此,我们认为远高于预期的值是信号。我们将基因g的原始信号定义为:
s[g,s]=c[g,s]-b[g,s],如果c[g,s]≥b[g,s]+2√b[g,s],否则为0。
因此,我们认为c[g,s]是真实信号,如果其大于该基因的背景水平平均值的两个标准偏差的话。
对各样品应用此程序将生成各样品中各基因的计数矩阵。我们还在此矩阵中包括来自以相同方式处理的h3k4me3chip的roadmapepigenomics数据的样品。
为了标准化不同覆盖的影响,我们认为“看家”常染色体基因的启动子处的信号在不同样品中应相似。我们将这些基因定义为一组参考健康样品中具有高度显著信号的基因。显著性水平的确切选择不改变标准化。
在原始信号样品x看家基因的矩阵上,我们应用了分位数标准化(bioconductornormalize.quantiles)。这导致各样品中看家基因的标准化值。但是,其没有为所有其他基因分配值。因此,我们估测各样品的乘法标准化因子,以使分位数标准化的值与原始值匹配最佳。对于大多数样品,两者之间的关系是线性的。
使比例因子重新缩放,以使参考健康样品集的总标准化信号(如下)为平均100万。
使用这些标准化因子,水平v[s],我们为各样品计算了标准化基因水平:
n[g,s]=v[s]*s[g,s]。
使用相同的标准化程序,我们还标准化了各样品中各窗口的覆盖率:
n[w,s]=v[s]*max(c[w,s]-b[w,s],0)。
定义组织特异性特征
使用roadmapepigenomics元数据表,我们定义了属于一个组织或一组组织的roadmap样品集。这些定义包括一些冗余。例如,淋巴细胞组包括b细胞、t细胞和nk样品,因此被归入这些组中的每一个。
然后,我们为各组定义了特定窗口组,在窗口w通过以下标准时:
1.窗口w在常染色体上;
2.在该组中的图谱样品中的至少一个中,n[w,s]≥35;
3.在该组外的所有图谱样品中,n[w,s]<15;
4.在w的1kb之内的所有窗口w'中,n[w,s]<15。
最后一个条件被加入是因为我们注意到,通常当基因被表达时,有“溢出(spillover)”到相邻窗口。
我们发现少于4个特定窗口的组被认为无特征。对于所有其他组,我们将特征定义为该组特定窗口(请参见表1)。cfchip处理和分析的工作流程被提供在图7中。
统计学检验
我们在这里使用两个主要检验:
检测检验。为了检验样品中在背景上存在基因还是特征,我们采用了泊松分布。更具体地:
计算检测p值(computedetectionpvalue)(w,s)
返回pλ(x≥x)ii泊松p-值
里w是一组窗口,其可以是与上述基因或组织特异性特征相关的窗口。
过度表达检验。为了检验一组基因的观察信号是否高于健康样品中的预期信号,我们利用健康对象的参考将基因的预期标准化信号h[g]限定为参考样品的n[g,s]平均值。
然后,我们使用以下程序:
计算过表达p值(computeoverexpressionpvalue)(g,s)
返回pλ(x≥x)ii|泊松p-值
以前的检验的主要区别在于,在我们从标准化单位转换为特定样品的单位后,我们纳入了健康样品的贡献。第二个区别是我们在基因水平上工作。
结果
实施例1:来自血浆的cf-核小体的染色质免疫沉淀
大多数血浆cfdna可能是具有完整组蛋白修饰的核小体dna(cf-核小体)形式。研究了从带有特定组蛋白标志物的cf-核小体中提取和测序dna是否可用于确定关于cfdna来源细胞的信息(图1a)。基于若干理由,这是受关注的方法。第一,通过设计的chip实验仅对阳性信号测序,减少了阳性信号所需的读段数目,从而降低了测定相关的成本和工作。第二,阳性目标相对稀少;启动子标志物,如h3k4me3,出现在基因组的~50,000个位置(小于基因组的1%)。增强子标志物(例如,h3k4me1)可出现在许多区域(基因组的~10%),但在各细胞中受限。第三,组蛋白标志物大部分是组织特异性的。具体地,大部分增强子是组织特异性的,因此提供强大的h3k4me1/3组织特异性(图1b)。第四,组蛋白修饰反映了转录活性并响应细胞状态的变化。因此,其创造了检测细胞其死亡时的活性变化的机会。
我们设计对于来自少至1-2ml血浆的cf-核小体chip-seq(cfchip)的简单方案(图1a插图,1c)。对来自11名健康个体的血浆样品进行的cfchip和配对末端测序关于h3k4me3和h3k4me1每个样品分别产生了30-170和90-2500万个独特读段,表明血浆中带有相应标志物(例如h3k4me3)的~1-2%的核小体被捕获,被接头连接,并被测序(参见材料和方法)。重要地,普遍表达的基因周围的cfchip信号显示出与来自组织的参考chip-seq(nihepigenomeroadmap协会)具有高度相关性(图1c)。在全球范围内,针对h3k4me1和h3k4me3的cfchip信号的元分析得出了这些标志物在增强子和启动子周围的预期一般分布(图1d)。
潜在的担忧是抽血过程中由白细胞溶解释放的染色质污染。几条证据表明这是高度不可能的。(a)cfchip文库的片段尺寸分布显示出~170和~320bp处对应于包裹在单核小体和双核小体周围的dna的两个峰(图1e),符合凋亡和在一些情况下坏死的细胞死亡,但不符合细胞溶解,产生10kb或更大的范围的片段。(b)我们鉴定了数千个携带h3k4me1的增强子和数十个携带h3k4me3的启动子,其不存在于构成有核血细胞最大部分的白细胞(外周血单核细胞;图1f-1g)的chip-seq中。对在cfchip中被h3k4me3标记但在白细胞中则不的启动子进行的分析确定了来自居于骨髓中的巨核细胞的强信号。(c)我们能够从患者的远处组织中检出疾病相关的染色质(见下文)。
非组蛋白dna缔合蛋白也可用于cf-chip。我们对人类血浆加标(spiked)90ng染色质——该染色质通过对来自小鼠胚胎干细胞的dna的天然mnase处理而制备,并用抗ctcf抗体进行cfchip。将cf-chip的测序读段与小鼠基因组进行比对,观察到清晰的与从小鼠细胞的ctcf的chip-seq获得的峰重叠的尖锐峰(图1h)。数据的元分析显示贯穿基因组的ctcf位点处的清晰信号,并且使用抗h3k4me3抗体的cfchip的类似分析显示,相同位点处组蛋白标志物耗尽(图1i)。ctcf结合和h3k4三甲基化总体上是互斥的,因此该结果有助于确认ctcf信号是真实的。
这些结果一起有力地表明,cf-核小体保留了活性组蛋白标志物和转录因子结合的完善建立的内源模式。我们在此点将分析集中于h3k4me3,因为将h3k4me3峰分配给特定基因相对简单直接。
为了评估cfchip的再现性,我们自多个对象中进行了技术和生物学复制。来自同一个体的复本和健康个体之间的复本分别显示0.94-0.97和0.92-0.94的相关性(图2a-c)。对象之间再现性低的峰富集x或y染色体特异性基因,并且在比较相同性别的两个个体时确实不明显(图2b)。
为了检验cfchip的检测限,我们利用了y染色体独特的序列,并将男性来源的血浆滴定到女性来源的血浆中。我们评价了具体基因组位置以及基因组特征的灵敏度——可以限定一定细胞类型或一定转录程序的差异表达的基因组位置的集合。y染色体上男性特异峰处的h3k4me3cfchip信号(图2d)显示,即使在男性血浆占据总血浆的小于10%,我们也可以可靠地鉴定出单个男性特异峰(图2d)。此外,男性特异峰的贡献随着男性血浆混合分数而线性地增加(图2e-f),证明cfchip灵敏度与该分数和特征位置的尺寸以及测序深度线性相关。事实上,将来自25个男性特异峰的信号组合可以将检测灵敏度提高到1%(图2e、2g)。这可能是低估值,因为二倍体基因组中有一个y染色体。从我们的男性加标实验推算,我们估测45至200个峰的适度特征尺寸可以在低测序深度下以高概率(0.95或更高)从构成cfdna库的0.1%的细胞检测cfdna(图2h)。这种尺寸的特征可以针对特定的细胞类型或转录程序进行识别(图2i)。
组织样品的启动子处的h3k4me3水平与转录水平相关,并有力地预测基因表达水平。我们发现cfchiph3k4me3与组成型基因处的白细胞rna-seq相关(基于roadmapepigenomicsconsortium和gtexconsortium2015),并且这种相关性与白细胞的chip-seq相似(图2j)。同样,我们发现与白细胞表达的基因的高度相关性与其对健康个体的cfdna库的主要贡献相一致(图2k)。这些结果有力表明,与转录相关的组蛋白修饰的cfchip可以提供对cfdna来源细胞的基因表达模式的了解。
考虑这些发现,我们着手检验检测健康对象的样品中组织特异性特征的能力。先前对cfdnacpg甲基化的研究估计,cfdna的~55%来自白细胞,并且~1%来自肝脏,而来自心脏和脑的cfdna贡献最小或没有。对多个健康组织使用h3k4me3chip-seq的roadmapepigenomics数据集作为参考,我们以无偏见的方式定义了组织特异性特征(参见材料和方法,表1)。然后,我们评价了各对象中各特征的标准化读段数目以及这些计数的统计学显著性(图2l-m;材料和方法)。如同预期,当使用几个特定峰或甚至单个特定峰时,可以检测到白细胞的存在。使用较大的特征,还清楚和显著地检测到肝cfdna的存在,相比之下大脑和心脏特征不存在于血液中,如同预期。这些信号是特异性的,并且具有高统计学置信度(q<10-20,参见材料和方法),并且其证明了cfchip检测来自稀少细胞群的cf-核小体的能力。
实施例2:cfchip检测与病理相关的细胞死亡。
cfchip识别来自远处组织的细胞的特征的能力表明令人兴奋的该工具检测源自疾病相关的病理性细胞死亡的cf-核小体的可能性。为了验证这一假设,我们从被诊断有急性心肌梗塞(ami)(一个导致广泛心肌细胞死亡的过程)的患者收集了样品。我们收集了进入急诊室的患者的样品,其中在紧急经皮冠状动脉介入(pci)以恢复血流之前、之后立即和之后~12小时获取样品。我们预期仅在ami患者中观察到心肌细胞死亡的cfchip信号,而在健康对象中观察不到,尤其是在pci后的样品中。
如预测,在pci后患者样品中强烈和显著检测到心脏特异性h3k4me3峰,但在健康个体的样品或pci前患者的样品中未检测到(图3a)。心脏信号包括在心脏特异性基因的启动子中的清晰的峰。例如,编码心脏特异性肌钙蛋白t2和i3的tnnt2和tnni3明显有活性,并且仅在这些样品中被观察到。这两个基因编码心肌损伤的典型蛋白质标记物(图3b)。事实上,我们发现在cfchip心脏特征的强度、血液中测得的肌钙蛋白水平、以及基于心脏特异性差异甲基化cpg的心脏cfdna估测之间的良好相关性(图3c)。
为了公平看待本发明描述来源组织的能力,我们评价了横跨cfchip样品的一组细胞类型特异性特征(图3d-e)。该分析表明,在所有样品中,我们都可以检测到来自血液(例如单核细胞和嗜中性白细胞)和器官(例如肝脏)的一定范围的细胞类型的特征。其中一名对象(h008)是怀有男胎的四个月孕妇,其样品呈现明显的胎盘特征。事实上,我们能够在其血浆中检测到低但显著的y染色体信号(图3d)。
在ami患者样品中,情况更为复杂。如上所述,pci后数小时采样的ami患者显示出清晰的心肌细胞特征(图3a、3d)。但是,此外,在pci之前和之后不久的ami患者中,我们观察到肝细胞特征显著增加。该特征包括肝脏特异性基因(如白蛋白和补体基因)的清晰信号(图3f)。这种出乎意料的观察结果可能是由于这样的公知现象:低器官灌注和肝缺氧继发的ami患者肝损伤。这些ami患者中的一个患者(m002)还呈现来自成红细胞相关基因的活性染色质水平升高,包括血红蛋白基因座(见下文)和促红细胞生成素基因座(epo),最有可能来自肝细胞——由于对缺氧的肝脏响应。这些数据一起表明对氧气缺乏的全身性响应。肝脏和成红细胞信号可能是由于与ami相关的全身灌注减少所造成的暂时性损伤。事实上,11个月后对该患者进行的跟进cfchip-seq呈现正常。在第二例ami患者(m001)中,我们观察到pci后数小时内肝特征逐渐下降,表明肝氧缺乏的迅速缓解(图3g)。为了确认我们的cfchip观察结果,我们分析了肝特异性基因的cfdna甲基化状态,这些基因的dna甲基化状态指示肝细胞死亡。事实上,我们观察到肝cfchip特征水平与肝cfdna估测值之间良好的一致性(r2=0.97,图3h)。
cfchip的重要潜在应用是癌症来源组织的鉴定。晚期癌症通常伴随着血浆中较高的cfdna含量,其中大部分来自肿瘤细胞(ctdna)。我们从胃肠道(gi)肿瘤患者收集血浆样品,并分析其cf-核小体来源组织(图3i、3e)。总体上,来自癌症患者的血浆包含在健康对象中观察不到的组织信号。最明显的是,我们观察到了源自胃肠道(gi)组织和胃肠道平滑肌的信号,这与肿瘤的原发部位一致。较弱但重要(显著,significant)的gi特征甚至在原发性肿瘤通过手术被去除并且仅残余转移性疾病明显时也是明显的(患者c004和c005)。我们还观察到来自其他组织的低而重要(显著,significant)的信号,例如在c001中观察到的脑信号。这些信号可能是由于治疗(c001接受了脑放疗)或由于对正常(非恶性)组织的附带损害所致。
我们还考察了接受部分肝切除术(phx)的局部化肝细胞癌(hcc)患者。我们在手术前、手术中和手术后的不同时间点采集了血液样品,并利用cfchip分析了循环cf-核小体,并测量了肝损伤经典标志物,酶alt(图3j)。惊人地,尽管事实是患者除hcc外还有活跃肝硬化,但术前alt水平也是正常的。alt水平在phx后的第一天上升,并在随后的几天期间逐渐下降。肝脏特征的cfchip分析与alt检验非常一致,再次表明cfchip检测到了远处组织的动态过程。一个区别是cfchip肝特征比alt早约2天降回到正常水平。这个区别可能是由于在循环中cfdna的半衰期(<2小时)比alt(~47小时)短。
总之,这些结果显示,与正在经历病理过程的患者相比,健康对象的cfchip信号存在明显差异。这些差异对应于发生这些过程发生的组织,如心脏、肝脏和胃肠道组织。
实施例3:血浆染色质反映基因活性模式
cfdna分析的主要挑战是推断来源组织中的基因表达。迄今为止,针对该问题提出的主要方法依靠cfdna中特定启动子元件的不足表达作为基因表达的指示。但是,这种方法需要极深的测序,并且限于目标组织的cfdna在血液中占主要群体的情况。我们测试了cfchip关于来源细胞中发生的非组成型基因表达程序可报告到什么程度。
h3k4me3与转录活性密切相关,并且响应转录程序的变化而动态变化,产生了cfchip可能能够在来源组织信息之外还检测更多动态转录程序的令人兴奋的可能性。为了验证这一假设,我们将来自ami或癌症患者的h3k4me3cfchip信号与代表不同细胞过程和响应的标志性基因表达特征集进行了比较(图4a-c)。
此分析发现多个特征具有高于预期的信号——即,针对该特征捕获的cf-核小体量显著高于我们在健康对象中的观察。例如,我们分别在经历缺氧和菌血症的患者m002和c005中观察到血红素代谢的强烈特征。c005的血细胞计数确实显示出高红细胞分布宽度(rdw)以及低血红细胞计数(rbc)和血红蛋白(hgb),表明由于贫血导致血红细胞高产。该信号可能是由于红细胞祖细胞或紧密相关的细胞的细胞死亡增加,或者由于成红细胞经历成熟变为红细胞时核丢失。因此,该特征指示特定的造血细胞分化过程。
其他特征(如糖酵解或干扰素-α响应)反映了可以在多种细胞类型中发生的过程。我们观察到癌症患者中较高的糖酵解特征,与被称为warburg效应的代谢重编程一致,这被认为是晚期癌症的标志(图4d)。我们还观察到遭受广泛肝损害的m002中的糖酵解特征明显增加。值得关注的是,在m002中,我们还观察到几种肝脏特异性糖酵解基因(如aldob和pfkfb1)的信号增加,而在癌症患者中,增强的糖酵解特征不包括来自这些基因的信号。这些结果表明,cfchip可以检测与潜在病理生理状态相关的细胞特异性转录程序。如预期,在癌症患者的血浆中,我们还观察到若干增殖相关特征(kras、myc靶标、e2f靶标、g2m检查点)增加以及协调代谢和细胞生长的mtorc1途径。值得关注的是,这些特征的部分在经历肝损害的ami患者中也被观察到,并且可能反映了pci后缺血性损害引起的肝脏恢复。
可检测的转录程序的另一实例是干扰素-α响应,该响应通常由于病原体例如病毒和细菌的存在而被引起。我们观察到m004和c005中的干扰素特征急剧增加。在后者中,这可能是由于导致其住院的严重菌血症。m004——其样品显示高的干扰素和炎症特征,与其他ami患者相比,就肌钙蛋白水平和cfchip心脏标志物而言似乎遭受了更严重的心脏损害(图3a、3c)。这可能是由于m004中引起了irf3/干扰素i响应,最近显示其促进严重的ami响应。
总之,这些观察结果显示,cf-核小体不仅报告特定细胞类型的死亡,而且还可以反映宽范围细胞类型中的基因表达程序的详细变化。
血浆染色质允许患者特异性分子表型的剥离(解剖,dissection)
癌细胞的标志是导致基因表达程序失调的遗传改变。这种癌症特异性转录程序的鉴定可以有助于诊断和治疗选择。对于各样品,我们测试了其信号与五个“参考”健康样品相比升高的基因。作为对照,参考组以外的无关健康样品与健康参考高度相关,其中极少基因(通常少于50个)显示显著升高的信号(图4e)。相比之下,来自患者的样品揭示了信号显著升高的数百至数千个基因(图4e)。针对在注释基因列表中的富集考察这些基因概括了上面讨论的一些结果。例如,针对gi道和脑的基因集富集c001中的基因,与该患者的病理学一致。
接下来,我们在h3k4me3cfchip信号中寻找癌症特异性特征。我们分析了来自癌症基因组图谱(thecancergenomeatlas)和gtex项目的表达谱,以关于各肿瘤类型鉴定肿瘤中与正常组织相比显著更高的基因集(参见材料和方法,表2)。然后,我们测试了样品中具有较高h3k4me3信号的基因集与在某肿瘤类型中过表达的基因集之间的显著重叠(参见材料和方法)。例如,c002与gi道腺癌基因有显著重叠(q<10-60)(图4e)。所有样品针对所有肿瘤类型的分析(图4f-g)显示,只有来自癌症患者的样品才具有肿瘤相关基因表达的显著富集,而健康人和mi患者则没有。重要地,该富集针对gi道癌症,与被诊断的病理一致。
关注已知在胃癌和结肠直肠癌中上调的特定基因,我们观察到与健康参考相比,这些患者中h3k4me3cfchip信号明显增加(图4h)。在这些基因中,我们发现了癌标志物ceacam5和ceacam6。这些基因的蛋白质产物用于临床癌症诊断的抗体基测定。第二种结肠直肠癌标志物,长的非编码rnaccat1(结肠直肠癌相关的转录体1),在其中一名癌症患者中显示强信号,但在健康对象中则不。另一个实例是长的非编码rnaegfr-as1,其介导癌症对egfr的成瘾,并且当被高度表达时可使肿瘤对egfr抑制不敏感。尽管在所有癌症中均检测到egfr的cfchip信号,但仅在c002中检测到egfr-as1,而在其他患者中未检测到。这一发现(该发现通过cfdna突变分析将检测不到)产生令人兴奋的可能性:cfchip可以在基因组突变以外还提供治疗选择信息。
表2:癌症特征
实施例4:增强子和基因体标志物的分析
我们对活性启动子标志物h3k4me3的分析提供了关于来源组织的转录程序的丰富信息。我们能从与增强子和基因活性相关的染色质标志物获得信息吗?h3赖氨酸4的单甲基化和二甲基化(分别为h3k4me1和h3k4me2)被发现于两种类型的基因组区域中:1)在标记有h3k4me3的区域边界处的启动子侧接区域,或2)平衡/活性增强子,其中h3k4me3几乎没有被检测到。组织中这些标志物的chip-seq显示在增强子附近的h3k4me2峰,而h3k4me1峰在增强子周围较宽(大约~10kb)。这些标志物的cfchip概括了预期的分布:在活性启动子周围,h3k4me2和h3k4me1位于h3k4me3主峰之侧(图5a-b),并且与h3k4me3和rna水平相关(图5d)。此外,在基因欠缺的区域,如ifnb1基因座中,我们清楚地看到增强子处的标志物,该标志物与实验验证的ifnb1增强子(banerjeeetal.2014)相匹配(图5c)。
由于h3k4me2更加凝缩并且健康对象之间信号高度相似(图5e),我们选择集中于此标志物以进行增强子分析。为了识别增强子,我们寻找了具有低h3k4me3信号或没有h3k4me3信号的h3k4me2峰。我们在健康对象中识别出~8,000个推定的增强子峰。这些峰中,基于多个组织中的五个染色质标志物(chromhmm),>90%位于预测为增强子区域的位点。此外,这些推定的增强子峰与相关细胞类型(例如单核细胞和嗜中性白细胞)中的chromhmm注释之间存在良好的一致性(图5c)。对来自结肠直肠癌患者的两个样品(c002.1和c002.2)应用相同的分析显示这些样品之间的强一致性,与健康对象具有实质性差异(图5e)。健康对象与癌症样品之间的差异说明了相对于启动子特征可从增强子获得的其他信息。癌症特异性增强子信号的几个实例包括tcf3、cdx1和ceacam5(图5f-h)。各测试对象中转录因子tcf3有启动子h3k4me3信号。然而,该基因附近的增强子活性标志物却截然不同,其中癌症样品显示在与推定的结肠增强子对应的区域中的清晰h3k4me2峰(图5f)。这些结果表明,该基因被细胞中对该信号有贡献的不同增强子激活,并且与患者的临床状况一致(图4a-g)。在成年人结肠中未观察到tcf3周围的h3k4me2峰的子集,仅在胎儿结肠中观察到,这与胎儿癌基因的去阻遏作用一致。在健康对象中,肠特异性转录因子cdx1没有h3k4me3信号。在癌症样品中,其具有清晰的信号。这种活性伴随着基因附近的大gi特异性增强子区域上的h3k4me2峰(图5g)。这表明cdx1通过这些增强子被激活。最后,考察ceacam5基因座,出现了更复杂的图谱(图5h)。在癌症样品中ceacam5具有在其启动子处的h3k4me3信号。这伴随着g1特异性增强子中的信号。然而,在健康对象中,在邻近增强子/启动子区域中存在强h3k4me2信号。这表明这些增强子中的一些可涉及单核细胞和嗜中性白细胞中ceacam5的阻遏。
在转录基因的主体内发现了h3赖氨酸36(h3k36me3)的三甲基化。不同于标记平衡和活跃基因处的转录起始位点的h3k4me3,h3k36me3需要活跃的转录伸长被布置(deposited),因此更能指示基因活性。h3k36me3的cfchip导致在基因体处的一般富集(图5i),并且信号与白细胞h3k36me3和rna-seq相关(图5j-k)。将健康对象的h3k36me3信号与结肠直肠腺癌患者的h3k36me3信号进行比较,我们发现在癌症样品中1500个基因的k36me3增加了2倍以上(图5l)。在这1500个基因中,已知60个基因在结肠腺癌中被上调(172个coad基因中的60个,p<10-20;图5l)。值得注意的是,尽管癌症中大多数具有较高h3k36me3基因体信号的基因也显示启动子处较高的h3k4me3,但我们发现了不一致的基因的实例(图5m),证明可以通过组合来自不同组蛋白标志物的数据获得更多信息。
总而言之,cf-chip-seq可以探测包括启动子、增强子和基因体在内的各种基因组功能性的状态,并且该信息对于来源细胞中的转录活性具有高度信息性。
实施例5:连续cfchip和用于ip的其他蛋白质。在cf-chip过程中,各血液样品的大多数物质都没有被捕获在珠上,因为其不携带目标修饰/蛋白质。以连续方式(其中在与一种抗体一起温育后将上清液传送给下一种)进行固定,大大提高了使用有限物料时的效率。即使经过多个cfchip步骤后,剩余材料(其仍然包含大部分原始cfdna)仍可用于基于dna的测定。为了测试可行性,进行h3k4me1然后h3k4me3的连续cfchip,反之亦然(图8)。发现这两个实验的结果之间有很好的一致性。
为了测试cf-chip靶向乙酰化组蛋白的能力,我们使用靶向h3上的不同乙酰化位点(h3k9ac、h3k27ac)和h2a组蛋白变体h2a.z乙酰化(h2a.z_ac)的抗体进行cf-chip。这些组蛋白标志物全部与活跃转录相关,并且事实上组蛋白乙酰化标志物显示在转录起始位点(tss)周围的一般富集模式。在cf-chip后,我们将测序的dna片段与人基因组进行比对,并围绕基因的tss为中心进行了元基因分析。如图9可见,所有乙酰基标志物均显示在tss周围的明显富集,如预期。我们还对结肠直肠癌(crc)患者使用了h3k27ac抗体,并且获得与健康供体相比远更高的信号,表明乙酰化组蛋白的cf-chip也可以具有诊断价值。
实施例6:通过邻近连接(proximityligation,mpl)chip进行的多重化
甚至比连续多轮chip更有效的是在同一管中进行所有抗体的固定。珠与抗体和dna接头结合,并带有该抗体的特异性匹配条形码。将具有不同抗体/条形码的珠在同一血样中混合,并在chip之后对所有珠进行连接反应。由于cfdna和接头/条形码的邻近和固相固定,反应是特异性的。各珠上的cfdna均用dna接头标记,该dna接头包含将其拉下的抗体的特异性条形码。对被任何蛋白质一起拉下的所有cfdna进行多重测序(图10a)。我们称这种方法为通过邻近连接(mpl)进行的多重化。此方法可最小化后续转移过程中的材料损失,并增加抗体在样品中找到其目标的机会。
为了证明mpl的可行性,我们证明了染色质可以被免疫沉淀在含有固定化抗体和条形码化dna接头的混合物的表面(mpl条形码化表面)上,可以生成固定在mpl条形码化表面上的染色质的下一代测序(ngs)dna文库,并且mpl条形码化表面之间的染色质混合最少。为了对此进行测试,设计了实验,如图10b所示。将独特的条形码与特定的下拉抗体(抗h3k4me3或抗h3k36me3)组合。将mpl条形码化的表面组合,以对源自两种酵母菌种(酿酒酵母和乳酸克鲁维酵母)的染色质进行chip,该两种酵母菌种可通过其基因组序列而被区分。为了测试在ip过程中混合是否发生,我们将k4和k36mpl条形码化表面与来自单一来源的染色质混合,并跟踪固定化染色质的基因组分布。k4mpl条形码化表面和k36mpl条形码化表面应分别使chip染色质偏向基因的5'或3'端。在ip步骤之后,我们还将与不同的酵母菌株温育的mpl条形码化表面进行组合,以测试所述方法中文库制备过程中和/或稍后的混合。在文库制备之前将ip的输出混合到单个试管中将表明在该阶段发生的混合,例如,在将乳酸克鲁维酵母dna用条形码1或条形码2测序的情况下,我们知道在文库制备过程中发生了不期望的混合。
我们用递减量的dna接头(并且因此递增量的蛋白g——其用于向mpl条形码化表面招募抗体)在chip后对mpl条形码化表面进行了qpcr,并计算了与输入相比固定化的染色质分数(图10c)。免疫沉淀的染色质分数与通过标准chip获得的结果相当,并且与固定化的条形码化dna接头量呈负相关(并且因此与固定化抗体的数量呈正相关)。注意,极低水平的背景信号(无蛋白g)证明了对mpl条形码化表面的chip依赖于抗体的固定化。
接下来,我们以含有抗h3k4me3或h3k36me3抗体的mpl条形码化表面进行chip-seq。测序后,将测序的dna片段与基因组进行比对,并将信号展示为沿一般基因的元(meta)作图(图10d)。具有h3k4me3和h3k36me3的mpl条形码化表面产生了典型的结果,其中h3k4me3集中在基因5'周围,而h3k36me3则在基因体周围向基因3'扩散。
最后,为了测试混合量,我们对从mpl条形码化表面获得的预期仅与5种不同样品中的乳酸克鲁维酵母染色质连接的读段数目进行计数。观察到大部分信号确实获自乳酸克鲁维酵母dna,而只有其余少部分获自酿酒酵母,表明即使在抗体未与mpl条形码化表面共价结合(结合由通过强生物素链霉亲和素相互作用与珠结合的生物素化蛋白g介导)的条件下混合也是最小限度的。事实上,混合的样品导致80-90%的与酿酒酵母的正确比对。
尽管已结合其具体实施方式描述了本发明,但是显然多种替代形式、改动和变型对于本领域技术人员将是显而易见的。因此,意图涵盖落入所附权利要求的精神和广泛范围内的所有这种替代形式、改动和变型。
- 还没有人留言评论。精彩留言会获得点赞!