一种用于治疗癌症的药物组合物的制作方法
本发明属于药物制剂领域,具体涉及一种含有n-(2-乙基-5-(6-(2-(三氟甲基)吡啶-3-基)咪唑并[1,2-b]哒嗪-2-基)苯基)-1-甲基环丙烷甲酰胺或其药理学上可接受的盐的药物组合物及其制备方法,该组合物具有溶出迅速、稳定性良好的特点。
背景技术:
hedgehog信号通路是一个重要的细胞生理过程调节通路。试验表明hedgehog通路对肿瘤的生长起着核心作用。因此,通过药物有效阻断hedgehog信号通路可抑制肿瘤细胞的生长,给恶性肿瘤的治疗提供了一条新的途径。
cn103261198a公开了一种小分子活性物质n-(2-乙基-5-(6-(2-(三氟甲基)吡啶-3-基)咪唑并[1,2-b]哒嗪-2-基)苯基)-1-甲基环丙烷甲酰胺,该活性物质的化学式如下式ⅰ所示:
该活性物质是口服型小分子hedgehog信号通路抑制剂,临床上可用于治疗转移性或局部晚期型基底细胞癌。
对于将难溶性药物制备成药物制剂的过程中,难溶性药物的溶出速度主要与制剂的崩解和溶蚀速度有关,而合适的粘合剂等药用辅料可以起到增加制剂溶出的有益效果。另外,在湿法制粒过程中,制软材是关键技术,软材的质量一般根据经验原则即“手握成团,轻压即散”来掌握,软材质量不合格容易出现成球或者粉末太多的现象,导致颗粒流动性较差,颗粒粒度分布不均匀,从而导致主药含量不均,影响药物的溶出。故粘合剂的使用在固体制剂的造粒工艺中起着关键性的作用。
预胶化淀粉,也叫做部分预胶化淀粉,较传统淀粉具有很大的优越性,它是利用加热和机械法使淀粉中的部分淀粉颗粒破裂成为一种胶状物,而另一部分仍维持原状态,并且严格控制胶化淀粉的比例,从而维持黏合与崩解性能的相对稳定。部分预胶化淀粉同时具有黏合和崩解的双重作用,其成本低于pvp和hpmc,可取代pvp、hpmc等化学合成辅料用于生产片剂与胶囊等固体制剂的粘合剂、崩解剂及填充剂。
在制备式i化合物的药物组合物的过程中我们发现,该化合物在各生理介质中均几乎不溶,属难溶性药物,采用常规的粘合剂,如淀粉浆溶液、交联聚维酮水溶液或羟丙甲纤维素水溶液制备组合物颗粒时,制粒过程成球严重,药物润湿性很差,制备的颗粒中大颗粒较多,颗粒流动性一般,不利于生产放大。
技术实现要素:
本发明的目的在于提供一种稳定性良好同时溶出迅速的药物组合物,并且该药物组合物制备工艺简单,更适合工业化大生产。
本发明提供的的药物组合物包括活性药物成分与预胶化淀粉。其中,活性药物成分为n-(2-乙基-5-(6-(2-(三氟甲基)吡啶-3-基)咪唑并[1,2-b]哒嗪-2-基)苯基)-1-甲基环丙烷甲酰胺或其药理学上可接受的盐,所述活性药物成分的含量范围可以是1-1000mg,优选25-500mg。预胶化淀粉的含量范围可以是基于组合物的总重量的1-90%,优选1-20%,更优选1-10%,最优选2-5%。
本发明提供的药物组合物还可以含有填充剂,例如微晶纤维素、磷酸氢钙、预胶化淀粉、乳糖、甘露醇等中的一种或多种。基于组合物的总重量,所述填充剂含量可以为约5-80%。
本发明提供的药物组合物还可以含有崩解剂,例如交联羧甲基纤维素钠、羧甲淀粉钠、低取代羟丙基纤维素或交联聚维酮中的一种或多种。基于组合物的总重量,所述崩解剂含量可以为约1-30%。
本发明提供的药物组合物还可包含一种或多种润滑剂,有助于压片或灌装胶囊。润滑剂包括滑石粉、硬脂酸镁、硬脂酸锌、硬脂富马酸钠、山嵛酸甘油酯、月桂醇硫酸钠、氢化植物油、胶体二氧化硅等。基于组合物的总重量,润滑剂的含量可以为约0.5-5%。
本发明的药物组合物中,所述活性成分的粒径分布范围d(0.9)可以小于100μm,优选小于50μm。
本发明还提供了一种药物组合物,包括:
1)1-70wt%的(n-(2-乙基-5-(6-(2-(三氟甲基)吡啶-3-基)咪唑并[1,2-b]哒嗪-2-基)苯基)-1-甲基环丙烷甲酰胺;
2)1-10wt%的预胶化淀粉;
3)5-80wt%的填充剂,所述填充剂选自乳糖、甘露醇和微晶纤维素中的一种或多种;
4)1-30wt%的崩解剂,所述崩解剂选自交联羧甲基纤维素钠、羧甲淀粉钠、低取代羟丙基纤维素和交联聚维酮中的一种或多种;
5)0.5-5wt%的润滑剂,所述润滑剂选自硬脂酸镁、硬脂富马酸钠、胶态二氧化硅和滑石粉的一种或多种。
优选地,本发明的药物组合物包括:
1)10-70wt%的(n-(2-乙基-5-(6-(2-(三氟甲基)吡啶-3-基)咪唑并[1,2-b]哒嗪-2-基)苯基)-1-甲基环丙烷甲酰胺;
2)2-5wt%的预胶化淀粉;
3)20-80wt%填充剂,所述填充剂包含微晶纤维素和乳糖;
4)4-8wt%的羧甲基淀粉钠;
5)0.5-2wt%的硬脂酸镁。
将本发明的药物组合物进行溶出测试,根据中国药典2010版二部附录溶出度测定第二法(桨法),使用2%十二烷基硫酸钠的0.1mol/l盐酸溶液作为溶出介质,对于本发明药物组合物的单位剂量,所述溶出介质优选为1000ml,并在37±0.5℃下以75rpm的桨速进行溶出试验,活性药物成分在45分钟内的溶出度可达到大于95%,优选在40分钟内的溶出度大于95%,更优选在35分钟内的溶出度大于95%,最优选在30分钟内的溶出度大于95%。
本发明的药物组合物中的预胶化淀粉优选作为粘合剂,预胶化淀粉可采用温度小于50℃,优选20-30℃的润湿剂配制成混悬液来制备药物组合物,润湿剂可以为水、乙醇或乙醇水溶液,优选水。相较于高温润湿剂配制的预胶化淀粉液,温度小于50℃的润湿剂配制的预胶化淀粉液在制备药物组合物过程中防止物料成球,增加颗粒流动性方面的效果更好。
本发明提供的药物组合物具有药物溶出迅速完全,稳定性好等优势,有利于工业化生产。辅料采用预胶化淀粉,可以明显改善制粒过程中物料润湿均一性差的现象,制备的颗粒均一性、流动性较好,颗粒压片后溶出迅速完全。
本发明的药物组合物具有优异的hedgehog信号通路抑制抑制活性,可用于与癌症有关的疾病,特别是转移性或局部晚期型基底细胞癌的治疗。
本发明还提供了一种制备药物组合物的方法,包括混合(n-(2-乙基-5-(6-(2-(三氟甲基)吡啶-3-基)咪唑并[1,2-b]哒嗪-2-基)苯基)-1-甲基环丙烷甲酰胺或其药理学上可接受的盐以及预胶化淀粉,可以采用本领域常见的方法制备成颗粒,例如高速剪切制粒、流化床一步制粒等方法制备药物组合物颗粒,然后压片(包衣)或灌装胶囊,制备片剂或胶囊剂。
附图说明
图1显示实施例1至5的片剂在2%sds的0.1mol/l盐酸溶液中的溶出曲线。
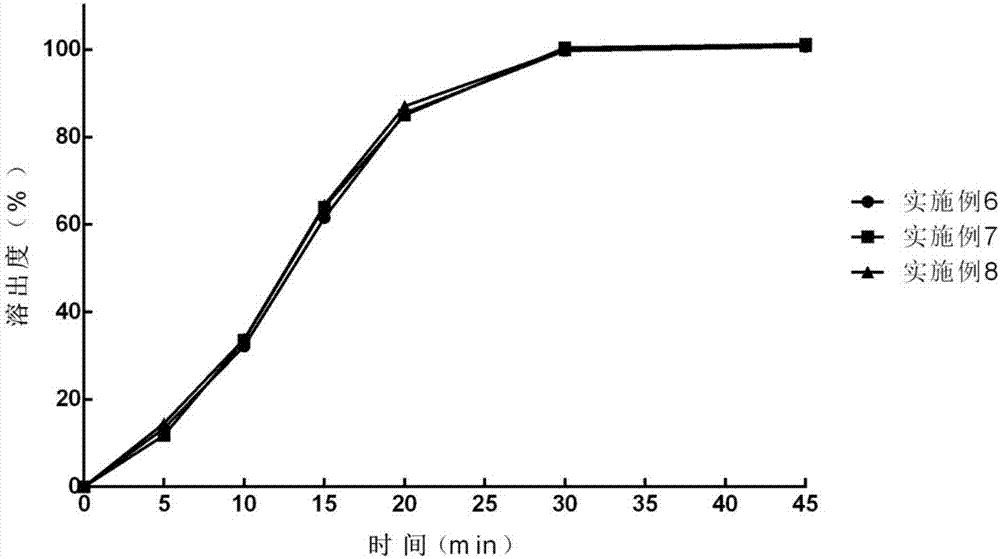
图2显示实施例6至8的片剂在2%sds的0.1mol/l盐酸溶液中的溶出曲线。
图3显示实施例9至11的片剂在2%sds的0.1mol/l盐酸溶液中的溶出曲线。
图4显示实施例12至14的片剂在2%sds的0.1mol/l盐酸溶液中的溶出曲线。
图5显示实施例15至20的片剂在2%sds的0.1mol/l盐酸溶液中的溶出曲线。
图6显示实施例6的片剂放置0个月,3个月,6个月后在2%sds的0.1mol/l盐酸溶液中的溶出曲线。
图7显示实施例10的片剂放置0个月,3个月,6个月后在2%sds的0.1mol/l盐酸溶液中的溶出曲线。
图8显示实施例14的片剂放置0个月,3个月,6个月后在2%sds的0.1mol/l盐酸溶液中的溶出曲线。
具体实施方式
通过以下实施例和实验例进一步详细说明本发明。这些实施例和实验例仅用于说明性目的,而并不用于限制本发明的范围。
实施例1至7
将粒径d(0.9)为45μm的n-(2-乙基-5-(6-(2-(三氟甲基)吡啶-3-基)咪唑并[1,2-b]哒嗪-2-基)苯基)-1-甲基环丙烷甲酰胺(以下简称为化合物a)、微晶纤维素、乳糖、羧甲淀粉钠,按表1中的比例进行混合,混合均匀后,分别以5%的羟丙甲纤维素水溶液、5%和8%的聚乙烯吡咯烷酮水溶液、5%和8%的淀粉浆溶液以及5%和8%的预胶化淀粉水溶液为粘合剂,采用高速剪切制粒机进行制粒,对软材进行整粒及干燥处理,然后将干颗粒(水分小于3%)进行干整粒,加入硬脂酸镁,混合。将得到的总混颗粒压片。制备实施例1~7的片剂。
表1
单位:g;规格:1000片
实验例1
观察实施例1~7中的干颗粒外观,并分别对其进行粒度分布测定。将45目筛放置于100目筛上侧,先称定物料总量,置45目筛上。保持水平状态过筛,左右往返,边筛动边拍打3min。取不能通过45目筛和能通过100目筛的颗粒,称定重量,计算其所占比例(%)。另外,分别测定其卡尔指数,结果如表2所示。
表2
表2的数据表明,选用hpmce5、聚乙烯吡咯烷酮-k30或淀粉浆溶液为粘合剂时,制粒过程成球严重,药物润湿性很差,制备的颗粒中大颗粒较多,颗粒流动性一般,加大粘合剂浓度,颗粒流动性及成球现象均无显著改善。而选用预胶化淀粉作为粘合剂,制粒过程成球现象得到改善,颗粒流动性显著提高。
实施例8~14
将粒径d(0.9)为45μm的化合物a、填充剂、崩解剂,按表3中的比例进行混合,混合均匀后,加入5%的预胶化淀粉水混悬液,采用高速剪切制粒机制粒,对软材进行整粒及干燥处理,然后将干颗粒(水分小于3%)进行干整粒,加入润滑剂,混合。将得到的总混颗粒压片。制备实施例8~14的片剂。
表3
单位:g;规格:1000片
实验例2
按实验例1中同样方法,对实施例8~14中的干颗粒分别进行粒度测定,并测定卡尔指数,结果如表4所示。
表4
表4的数据表明,使用预胶化淀粉作为粘合剂的实施例8~14的片剂,均能较好的改善制粒过程中出现的成球现象,制得的颗粒粒度均匀,流动性良好,改变其他赋形剂的种类,影响不大。
实验例3:溶出实验
根据中国药典2015版二部附录溶出度测定第二法(桨法),分别对实施例1~5、6~8、9~11、12~14的片剂进行溶出度测定。使用2%十二烷基硫酸钠(sds)的0.1mol/l盐酸溶液1000ml作为溶出介质,并在37±0.5℃下以75rpm的桨速进行溶出试验。结果表明,使用羟丙甲纤维素、聚乙烯吡咯烷酮-k30或淀粉浆溶液为粘合剂的实施例1~5的片剂,化合物a在45min时溶出仅80%左右,溶出不完全,溶出曲线图见图1;使用预胶化淀粉冷配后的混和液作为粘合剂的实施例6~14的片剂,化合物a的溶出均迅速且完全,溶出曲线图见图2~4。
实施例15~20
将不同粒度d(0.9)的化合物a、微晶纤维素、乳糖、羧甲淀粉钠,按表5中的比例进行混合,混合均匀后,加入5%的预胶化淀粉水混悬液,采用高速剪切制粒机进行制粒,对软材进行湿整粒及干燥处理,然后将干颗粒(水分小于3%)进行干整粒,加入硬脂酸镁,混合。将得到的总混颗粒压片。制备实施例15~20的片剂。
表5
单位:g;规格:1000片
实验例4:溶出实验
根据中国药典2015版二部附录溶出度测定第二法(桨法),对实施例15~20的片剂进行溶出度测定。使用2%sds的0.1mol/l盐酸溶液1000ml作为溶出介质,并在37±0.5℃下以75rpm的桨速进行溶出试验。结果表明,化合物a粒度(d0.9)在100μm以下时,均能在45min内溶出完全。具体来说,实施例15~17的片剂,粒度在50μm以下时,化合物a的溶出迅速完全;实施例18~19的片剂,粒度在50μm~100μm之间,与实施例15~17相比,化合物a的溶出完全,但速度减慢。而实施例20的片剂,粒度在100μm以上时,化合物a在45min时溶出仅90%左右,溶出不完全,溶出曲线图见图5。
实验例5:稳定性研究
将实施例6、10、14中的片剂,置于40℃±2℃、相对湿度75%±5%的环境下,分别于0个月,3个月,6个月取样,观察其性状,采用hplc法测定其有关物质和含量。根据中国药典2015版二部附录溶出度测定第二法(桨法),使用2%sds的0.1mol/l盐酸溶液1000ml作为溶出介质,并在37±0.5℃下以75rpm的桨速分别对其进行溶出度测定。
有关物质和含量测定结果表明(表6至8),在0至6个月加速条件考察期间,实施例6、10、14中的片剂其性状、含量和有关物质无显著变化,样品稳定。
溶出度测定结果表明,实施例6、10、14中的片剂中化合物a的溶出度在加速条件下放置0个月,3个月,6个月后仍没有明显下降,溶出度均迅速且完全(见图6至8)。
表6实施例6的有关物质和含量测定结果
表7实施例10的有关物质和含量测定结果
表8实施例14的有关物质和含量测定结果
- 还没有人留言评论。精彩留言会获得点赞!