DNA混合物中的组织甲基化模式分析的制作方法
本申请要求以下临时申请的优先权且是以下临时申请的正式申请:Chiu等人在2014年7月18日提交的名称为“通过组织特异性甲基化模式分析来测定DNA混合物的组成(DeterminingtheCompositionsofaDNAMixturebyTissue-SpecificMethylationPatternAnalysis)”的美国临时申请62/026,330;Chiu等人在2015年5月7日提交的名称为“通过组织特异性甲基化模式分析来测定DNA混合物的组成”的美国临时申请62/158,466;以及Chiu等人在2014年6月23日提交的名称为“通过组织特异性甲基化模式分析来测定DNA混合物的组成”的美国临时申请62/183,669,所述临时申请均整体并入本申请供参考以用于所有目的。本申请还与名称为“无创测定胎儿或血浆肿瘤的甲基化组(Non-InvasiveDeterminationOfMethylomeOfFetusOrTumorFromPlasma)”的共同拥有的PCT公开WO2014/043763相关,所述公开整体并入本申请供参考以用于所有目的。
背景技术:
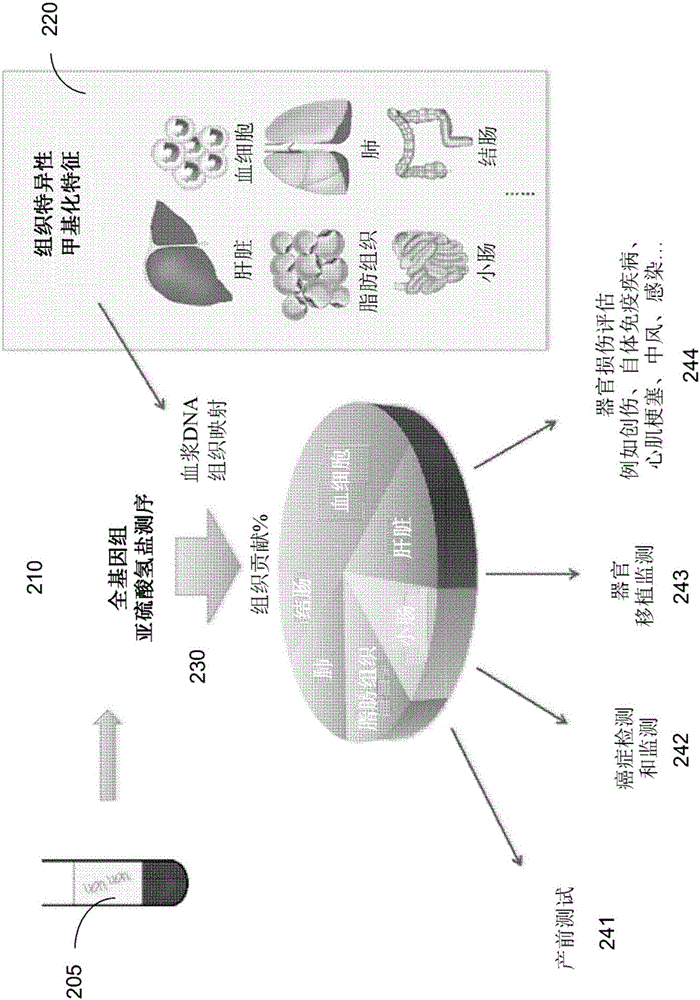
:分析血浆中的游离DNA已经表明适用于不同的诊断目的,包括无创产前测试和癌症检测。人们相信,血浆中存在游离DNA是因为DNA从凋亡的细胞中释放出来(Jahr等人,《癌症研究(CancerRes)》2001;61:1659-1665和Lo等人,《科学·转化医学(SciTranslMed.)》2010;2:61ra91.)。在此前的研究中,已经表明造血细胞是健康个体和器官移植接受者中血浆DNA的主要来源(LuiYY等人,《临床化学(ClinChem)》2002;48:421-7和ZhengYW等人,《临床化学》2012;58:549-58)。在这些此前研究中,使用器官移植模型测定不同器官对血浆DNA的贡献。在那些情形中,利用器官供者与移植接受者之间的遗传学差异来计算所移植器官对移植接受者的血浆DNA的贡献。然而,在这种模型中,只能测定所移植器官的贡献,而来自接受者的其它器官的贡献无法同时测定。另外,即使存在可以利用甲基化模式测定其它器官的贡献的技术,这种技术的准确性也尚未受到全面测试,且因此尚未充分鉴定准确性方面的缺陷。并且,其它器官贡献的测定应用已经受到限制。技术实现要素:所述实施例测定了不同组织对生物样品的贡献,所述生物样品含有来自不同组织类型的游离DNA分子的混合物,例如血浆和其它体液中所出现。实施例可以分析DNA混合物的甲基化模式(例如特定基因组位点的甲基化水平)且测定不同组织类型对DNA混合物的贡献率。可以选择具有跨越组织类型和跨越个体的特定性质的各种类型的基因组位点,以便在测定各种组织类型的贡献时提高准确性。举例来说,可以使用变异率至少达到阈值量的基因组位点;与此相反的是,只使用一种组织类型特有的基因组位点。在一些实施例中,可以测定对DNA混合物具有潜在贡献的组织类型(候选组织)的甲基化模式。然后测定所关注的DNA混合物的甲基化模式。举例来说,可以计算不同位点的甲基化水平。由于DNA混合物是由来自候选组织的DNA组成,因此可以通过比较DNA混合物的甲基化模式和候选组织类型来测定DNA混合物的组成。举例来说,可以利用N个基因组位点的甲基化水平计算M个组织的贡献,其中M小于或等于N。可以针对各种组织每个位点的甲基化水平。可以对线性方程式体系Ax=b求解,其中b是在N个位点所测量的甲基化密度的向量,x是M个组织的贡献的向量,且A是M个行和N个列的矩阵,其中每一行在那一行的特定位点处提供N个组织的甲基化密度。如果M小于N,那么可以进行最小二乘方优化。在各种实施例中,特定组织类型对DNA混合物的贡献百分率相对于参考值的显著分离值(即,减去的差值或比率)可以指示患病状态。参考值可以对应于健康个人中所测定的贡献百分率,且分离值大于阈值可以确定患病状态,因为病变组织释放的游离DNA分子多于健康组织。在其它实施例中,可以是利用两组游离DNA分子的甲基化水平测定组织类型的两种贡献率,每一组针对不同尺寸范围,以分类鉴定组织类型是否患病。可以将两种贡献率之间的分离值与阈值进行比较,且可以基于比较来分类确定第一组织类型是否处于疾病状态。举例来说,这种技术可以通过测量对较短游离DNA分子的较高贡献率(高于较长游离DNA分子)来鉴定释放较短游离DNA分子的病变组织。在又其它实施例中,可以利用两组游离DNA分子的甲基化水平测定一种组织类型的两种贡献率,每一组针对不同染色体区域,以分类鉴定第一染色体区域是否具有序列不均衡。可以对两种贡献率之间的分离值与阈值进行比较,且可以基于比较来分类确定第一染色体区域是否具有序列不均衡。举例来说,拷贝数不同的区域将对应于组织类型不同的贡献百分率,这是拷贝数失常的起因,因为当组织类型具有有失常的肿瘤时,这可能会发生。其它实施例涉及与本文所述方法有关的系统和计算机可读媒体。参照以下具体实施方式和附图可以更好地了解本发明实施例的性质及优点。附图说明图1是一个流程图,其图解说明根据本发明的实施例分析游离DNA分子的DNA混合物以利用甲基化水平测定不同组织类型的贡献率的方法。图2绘示一个示意图,其根据本发明的实施例绘示了DNA甲基化解卷积的若干潜在应用(例如使用血浆)和其应用。图3A根据本发明的实施例绘示了15位孕妇的不同器官对血浆DNA的贡献百分率图。图3B根据本发明的实施例绘示了图350,其是根据血浆DNA甲基化解卷积所推知的胎盘所贡献的血浆DNA分率与使用胎儿特异性SNP等位基因所推知的胎儿DNA分率之间的相关性。图4根据本发明的实施例绘示了利用孕妇间的血浆DNA组织映射分析所测定的贡献百分率表。图5根据本发明的实施例、依据血浆DNA组织映射和基于胎儿特异性SNP等位基因的胎儿DNA分率绘示了除胎盘之外的器官的贡献百分率图。图6根据本发明的实施例绘示了得自非怀孕健康对照个体间的血浆DNA组织映射分析的贡献百分率表。图7根据本发明的实施例绘示了使用第一组标记(器官特异性较高)估算的11位孕妇和4位非怀孕健康个体的不同器官对血浆DNA的贡献表。图8根据本发明的实施例绘示了使用第二组标记(器官特异性较低)估算的11位孕妇和4位非怀孕健康个体的不同器官对血浆DNA的贡献表。图9A图示了所估算的胎儿DNA分率(胎盘的贡献)与通过母体血浆样品中的胎儿特异性等位基因计数所测定的胎儿DNA分率之间的相关性。图9B图示了利用甲基化标记的估算与通过胎儿特异性等位基因计数所测定的胎儿DNA分率之间的绝对差。图10根据本发明的实施例、基于器官特异性甲基化模式分析绘示了癌症患者和健康患者的不同组织对血浆DNA的贡献表1000。图11A是图1100,其根据本发明的实施例绘示了通过器官特异性甲基化模式分析所测定和根据全基因组甲基化水平所测定的肿瘤DNA分率值。图11B图示了肝脏所贡献的血浆DNA分率(基于血浆DNA组织映射分析)与通过GAAL分析所测定的肿瘤源血浆DNA分率之间的相关性。图12A图示了患者HCC10在不同时间的血浆中的估算肿瘤源DNA。图12B图示了患者HCC9血浆中的估算肿瘤源DNA。图13是一个表,其根据本发明的实施例绘示器官移植患者间的血浆DNA组织映射分析。图14图示了根据血浆DNA组织映射所推知的移植物所贡献的血浆DNA分率与使用供者特异性SNP等位基因所测定的供者DNA分率之间的相关性。图15A图示了一种分析,其对使用503个I型、503个II型以及两种类型(各503个)标记进行甲基化解卷积的准确性进行了比较。图15B图示了一种分析,其对使用251个I型、251个II型以及两种类型(各251个)标记进行甲基化解卷积的准确性进行了比较。图16A图示了一种分析,其对使用123个I型、123个II型以及两种类型(各123个)标记进行甲基化解卷积的准确性进行了比较。图16B图示了一种分析,其对使用52个I型、52个II型以及两种类型(各52个)标记进行甲基化解卷积的准确性进行了比较。图17A图示了一种分析,其对使用26个I型、26个II型以及两种类型(各26个)标记进行甲基化解卷积的准确性进行了比较。图17B图示了一种分析,其对使用13个I型、13个II型以及两种类型(各13个)标记进行甲基化解卷积的准确性进行了比较。图18A根据本发明的实施例图示了使用具有不同选择准则的标记所推知的胎盘对血浆DNA的贡献。图18B图示了在同类型组织中使用具有低变异率(i类)和高变异率(ii类)的标记进行血浆DNA解卷积的准确性。图19是一个表,其根据本发明的实施例、基于器官特异性甲基化模式分析绘示了各种癌症患者和健康个体的不同组织对血浆DNA的贡献。图20绘示了一个表,所述表根据本发明的实施例绘示了每位癌症患者的不同器官的贡献与四位对照个体的平均值的比较情况。图21A根据本发明的实施例图示了利用甲基化标记所估算的HCC和健康对照个体的肝脏对血浆DNA的贡献。图21B图示了健康对照者与HCC患者之间,肝脏对血浆DNA的贡献百分率,如根据本发明的实施例所推知。图22A和22B绘示了根据本发明的实施例、通过非怀孕健康对照者与肺癌或结肠直肠癌患者之间的比较所推知的(A)肺和(B)结肠的贡献百分率。图23是一个表,其根据本发明的实施例绘示了癌症患者间的血浆DNA组织映射分析。图24是一个流程图,其根据本发明的实施例图解说明了分析游离DNA分子的DNA混合物的方法,以基于组织对DNA混合物的贡献率提高来鉴定所述组织的疾病状态。图25是一个表,其根据本发明的实施例绘示了通过甲基化解卷积而得到的九位SLE患者的不同器官对血浆DNA的贡献百分率。图26A根据本发明的实施例图示了所测定的三位孕妇(M6941p、M7171p和M396p)的胎盘对不同长度的游离DNA分子的贡献。图26B是一个表,其根据本发明的实施例绘示了所测定的移植患者的非造血组织对不同长度的游离DNA分子的贡献。图27A根据本发明的实施例图示了所测定的移植患者的肝脏对不同长度的游离DNA分子的贡献。图27B根据本发明的实施例图示了所测定的HCC患者的肝脏对不同长度的游离DNA分子的贡献。图28是一个流程图,其根据本发明的实施例图解说明了一种分析游离DNA分子的DNA混合物的方法,以基于组织对不同尺寸的游离DNA分子的DNA混合物的差异贡献率来鉴定所述组织的疾病状态。图29是一个流程图,其根据本发明的实施例图解说明了一种用于确定拷贝数失常的起源组织的方法2900。图30A根据本发明的实施例绘示了携有第21对染色体三体症的孕妇的染色体特异性血浆DNA甲基化解卷积的分析的图解说明。图30B是图表3050,其根据本发明的实施例绘示了各怀有第21对染色体三体症(T21)胎儿的孕妇的不同组织间的染色体21的分离值ΔM。图31是一个图表,其根据本发明的实施例绘示了各怀有第21对染色体三体症(T21)胎儿的孕妇的不同组织间的其它染色体的分离值ΔM。图32A根据本发明的实施例图解说明了癌症患者的血浆DNA的CNA区域的分析。图32B是一个图表,其根据本发明的实施例绘示了癌症患者的不同组织间的展现拷贝数增加的区域与展现拷贝数损失的区域之间的分离值ΔM。图33是一个图表,其根据本发明的实施例绘示了癌症患者的不同组织间的随机选择基因组区域之间的分离值ΔM。图34A根据本发明的实施例绘示了患有并发淋巴瘤的孕妇的甲基化解卷积分析的图解说明。图34B图示了从孕妇所收集到的试样间的拷贝数失常检测的全基因组DNA测序分析,所述孕妇经诊断在怀孕早期期间患有复发滤泡性淋巴瘤。图35A是表3500,其绘示了使用患有复发滤泡性淋巴瘤的孕妇的治疗前血浆样品,根据血浆DNA组织映射所测定的贡献率。图35B是一个图表,其绘示了患有并发滤泡性淋巴瘤的孕妇的不同组织的分离值ΔM。图36A图示了结肠直肠癌转移到肝脏的患者的血浆DNA的拷贝数失常分析。图36B是一个图表,其根据本发明的实施例绘示了患有结肠直肠癌和肝脏转移的患者的血浆DNA的拷贝数失常的甲基化解卷积分析。图37和38绘示了各种样品的基本测序参数表,包括测序深度,其用于鉴定起源组织。图39是一个流程图,其根据本发明的实施例图解说明了一种使用甲基化解卷积来分析生物体的生物样品的方法,以确定染色体区域是否展现序列不均衡。图40A根据本发明的实施例图示了两位孕妇的尿液DNA的尺寸分布。图40B根据本发明的实施例绘示了尿液DNA中的不同染色体的基因组表示(GR)图。图41绘示了可配合根据本发明实施例的系统和方法使用的计算机系统10实例的方块图。附录A绘示了I型和II型标记的表S1。具体实施方式术语“甲基化组”提供了基因组中多个位点或基因座的DNA甲基化数量的量度。甲基化组可以对应于基因组的全部、基因组的大部分或基因组的相对较小部分。“胎儿甲基化组”对应于怀孕女性胎儿的甲基化组。胎儿甲基化组可以使用多种胎儿组织或胎儿DNA来源(包括胎盘组织和母体血浆中的胎儿游离DNA)来测定。“肿瘤甲基化组”对应于生物体(例如人类)的肿瘤甲基化组。肿瘤甲基化组可以使用肿瘤组织或母体血浆中的肿瘤游离DNA测定。胎儿甲基化组和肿瘤甲基化组是所关注的甲基化组的实例。所关注的甲基化组的其它实例是器官的甲基化组(例如脑细胞、骨、肺、心脏、肌肉和肾脏等的甲基化组),所述甲基化组可以促进DNA进入体液(例如血浆、血清、汗液、唾液、尿液、生殖器分泌物、精液、便液、腹泻液、脑脊髓液、胃肠道分泌物、腹水液、胸膜液、眼内液、水囊肿液(例如睾丸)、囊肿液、胰腺分泌物、肠分泌物、痰液、泪液、乳房和甲状腺的抽吸液等)。器官可以是移植的器官。“血浆甲基化组”是从动物(例如人类)血浆或血清中测定的甲基化组。血浆甲基化组是游离甲基化组的一个实例,因为血浆和血清包含游离DNA。由于血浆甲基化组是胎儿/母体甲基化组或肿瘤/患者甲基化组或来源于不同组织或器官的DNA的混合物,因此血浆甲基化组也是混合型甲基化组的一个实例。“胎盘甲基化组”可以利用绒毛样品(CVS)或胎盘组织样品(例如分娩后获得)测定。“细胞甲基化组”对应于从患者的细胞(例如血细胞)测定的甲基化组。血细胞的甲基化组称为血细胞甲基化组(或血液甲基化组)。“位点”对应于单个位点,其可以是单个碱基位置或一组相关碱基位置,例如CpG位点。“基因座”可以对应于包括多个位点的区域。基因座可以包括仅一个位点,这使得所述基因座在那个背景下相当于一个位点。每个基因组位点(例如CpG位点)的“甲基化指数”是指在所述位点上展示甲基化的序列读数占覆盖那个位点的读数总数的比例。区域的“甲基化密度”是区域内展示甲基化的位点的读数数目除以所述区域中覆盖所述位点的读数总数。位点可以具有具体的特征,例如是CpG位点。因此,区域的“CpG甲基化密度”是展示CpG甲基化的读数数目除以覆盖所述区域中CpG位点(例如特定CpG位点、CpG岛内的CpG位点或较大区域)的读数总数。举例来说,人类基因组中每个100kb分组的甲基化密度可以利用亚硫酸氢盐处理之后的CpG位点上未转化的胞嘧啶(其对应于甲基化胞嘧啶)总数占对应于100kb区域的序列读数所覆盖的所有CpG位点的比例来测定。这种分析也可以根据例如50kb或1Mb等其它分组尺寸进行。区域可以是全基因组或染色体或染色体的一部分(例如染色体臂)。当一个区域仅含一个CpG位点时,那个CpG位点的甲基化指数与所述区域的甲基化密度相同。“甲基化胞嘧啶的比例”是指所述区域中,按所分析的胞嘧啶残基(即,包括CpG背景外部的胞嘧啶)总数计,展示甲基化(例如在亚硫酸氢盐转化之后未转化)的胞嘧啶位点“C”的数目。甲基化指数、甲基化密度和甲基化胞嘧啶的比例是“甲基化水平”的实例。“甲基化特征”(也称为甲基化状态)包括与区域的DNA甲基化有关的信息。与DNA甲基化有关的信息可以包括(但不限于)CpG位点的甲基化指数、区域中CpG位点的甲基化密度、相邻区域上CpG位点的分布、含有超过一个CpG位点的区域内每一个别CpG位点的甲基化模式或水平,以及非CpG甲基化。基因组中大部分的甲基化特征可以视为相当于甲基化组。哺乳动物基因组中的“DNA甲基化”通常是指甲基添加到CpG二核苷酸间的胞嘧啶残基的5'碳(即5-甲基胞嘧啶)。DNA甲基化可以发生于其它背景下的胞嘧啶中,例如CHG和CHH,其中H是腺嘌呤、胞嘧啶或胸腺嘧啶。胞嘧啶甲基化还可以呈5-羟甲基胞嘧啶形式。还报道了非胞嘧啶甲基化,例如N6-甲基腺嘌呤。“组织”对应于相同类型的一组细胞。不同类型的组织不仅可以由不同类型的细胞(例如肝细胞、肺泡细胞或血细胞)组成,而且可以对应于来自不同生物体(母亲相对于胎儿)的组织或对应于健康细胞相对于肿瘤细胞。“参考组织”对应于测定组织特异性甲基化水平所用的组织。可以利用来自不同个体的相同组织类型的多个样品测定那种组织类型的组织特异性甲基化水平。「生物样品」是指取自个体(例如人类,例如孕妇、癌症患者或怀疑患有癌症者、器官移植接受者或怀疑患有涉及器官(例如心肌梗塞的心脏或中风的脑)的疾病过程的个体)并且含有一或多个所关注的核酸分子的任何样品。生物样品可以是体液,诸如血液、血浆、血清、尿液、阴道液、水囊肿液(例如睾丸),或阴道冲洗液、胸膜液、腹水液、脑脊髓液、唾液、汗液、泪液、痰液、支气管肺泡灌洗液等。也可以使用粪便样品。术语“癌症等级”可以指癌症是否存在、癌症阶段、肿瘤尺寸、是否存在转移、身体的总肿瘤负荷,和/或癌症严重程度的其它量度。癌症等级可以是数字或其它标志,诸如符号、字母和颜色。等级可以是零。癌症等级还包括与突变或突变数目相关的恶化前或癌变前病状(状态)。癌症等级可以按各种方式使用。举例来说,筛选可以检查已知此前未患有癌症的某人是否存在癌症。评估可以调查已经诊断患有癌症的某人以监测癌症随时间的进展、研究疗法的有效性或确定预后。在一个实施例中,预后可以表示为患者死于癌症的机率,或在具体持续期或时间之后癌症进展的机率,或癌症转移的机率。检测可以意指‘筛选’或可以意指检查具有癌症的潜在特征(例如症状或其它阳性测试)的某人是否患有癌症。术语染色体区域的“序列不均衡”意指染色体区域中的游离DNA分子数量相对于期望值(如果生物体健康)的任何显著偏离。举例来说,某个组织中的染色体区域可以展现扩增或缺失,从而导致含有所述组织DNA与其它组织DNA混合的DNA混合物中出现染色体区域的序列不均衡。举例来说,期望值可以获自另一个样品或获自假设正常的另一个染色体区域(例如代表二倍体生物体的两个拷贝的数量)。染色体区域可以由多个不相交的子区域组成。基因组基因座的“类型”(标记)对应于跨越组织类型的基因座的具体属性。描述主要指I型基因座和II型基因座,其性质详细提供于下文中。给定类型的基因座在跨越组织类型的甲基化水平上可以具有具体的统计变异。基因组基因座的“类别”(标记)对应于相同组织类型的基因座的甲基化水平在不同个体间的的具体变异。一组基因组基因座(标记)可以由任意数目个不同类型和/或类别的基因座组成。因此,一组基因座对应于针对特定测量手段所选择的基因座且不意指所述组中的基因座的任何特定性质。“分离值”对应于涉及两个值(例如两个贡献率或两个甲基化水平)的差值或比率。分离值可以是简单的差值或比率。分离值可以包括其它因数,例如相乘因数。作为其它实例,可以使用所述值的函数的差值或比率,例如两个值的自然对数(ln)的差值。如在此所用,术语“分类”是指与样品的特定性质有关的任何数字或其它字符。举例来说,“+”符号(或词语“阳性”)可以表示样品归类为具有缺失或扩增。分类可以是二元的(例如阳性或阴性)或是更多等级的分类(例如1到10或0到1的量表)。术语“截止值”和“阈值”是指运算中所用的预定数字。举例来说,截止尺寸可以是指一种尺寸,高于所述尺寸则排除片段。阈值可以是一种值,高于或低于所述值则适用特定分类。这些术语中的任一个可以在这些背景中的任一背景下使用。详细描述本发明的实施例可以利用特定组织类型在某些基因组位点的已知甲基化水平来测定各种组织类型的游离DNA(或其它DNA混合物)在血浆中的百分比。举例来说,可以测量肝脏样品在基因组位点的甲基化水平,且这些组织特异性甲基化水平可以用于测定混合物中有多少游离DNA是来自肝脏。还可以针对向DNA混合物提供实质性贡献的组织类型来测量甲基化水平,以便可以说明游离DNA混合物的优势(例如大于90%、95%或99%)。此类其它样品可以包括(但不限于)以下中的一些或全部:肺、结肠、小肠、胰脏、肾上腺、食道、脂肪组织、心脏和脑。解卷积方法可以用于测定组织特异性甲基化水平已知的每种组织类型的贡献率(例如百分比)。在一些实施例中,可以利用指定基因组位点的已知组织特异性甲基化水平和混合甲基化水平创建线性方程式体系,且可以测定最接近于所测量的混合甲基化水平的贡献率(例如利用最小二乘方)。可以选择特定的基因组位点以提供所期望的准确度。举例来说,可以使用变异率至少达到阈值量的基因组位点;与此相反的是,只使用一种组织类型特有的基因组位点。可以选择第一组(例如10个)基因组位点,使得每个位点在组织类型间具有至少0.15的甲基化水平变异系数且使得M个组织类型的每个位点的最大与最小甲基化水平之间的差异在一或多个其它样品中超过0.1。此第一组基因组位点可能不具有特定组织类型的特定甲基化标志,例如仅在或主要在特定组织类型中甲基化。此第一组称为II型位点。这些基因组位点可以与具有特定标志的基因组位点(称为I型位点)组合使用。使用II型位点可以确保基因组位点跨越甲基化水平在组织类型间的完整空间,借此提高I型位点上的准确性。仅使用更多I型位点向甲基化空间提供冗余的基本向量(即,更多的基因组位点与其它位点具有相同模式),而增添甲基化水平在不同组织间具有不同值的其它基因组位点为经由线性方程式体系鉴别贡献率增添了新的基本向量。测定贡献率后(不论所选位点的类型),贡献率便可以用于各种目的。各种组织类型的参考贡献率可以针对就那些组织类型来说健康的特定人群加以测定(例如所有组织类型健康个体或某些组织类型健康个体)。当一种组织类型(例如肝脏)发生病变时,那么那种组织会释放更多的游离DNA分子,如可以经由细胞凋亡发生。举例来说,肝脏的贡献率的实质性增加(即,阈值大于参考值)表明肝脏发生病变。可以进一步分析特定组织类型的贡献率的此类增加,例如游离DNA的尺寸分析。尺寸分析也可以单独进行。两种贡献率可以根据不同尺寸范围(例如短和长)测定,且两种贡献率之间的分离(即,差异或比率)可以表明来自特定组织类型的短游离DNA分子比长游离DNA分子多。由于病变组织具有较短的游离DNA分子,因此特定组织类型对较短游离DNA分子的贡献率高于较长游离DNA分子表明特定组织类型发生病变。使用不同染色体区域的组织类型的贡献率之间的分离可以用于确定所述组织类型是否具有序列不均衡。在所述组织类型是胎儿组织的怀孕女性的一个实例中,如果染色体21存在三个拷贝,那么胎儿组织的百分比经测量(使用来自染色体21的游离DNA)高于具有两个拷贝的另一个染色体。胎儿组织的贡献率的显著分离(例如大于阈值)表明染色体21具有序列不均衡。作为检测序列不平衡的另一实例,可以鉴定出具有拷贝数失常的特定染色体区域,但失常的起因可能未知。也可以怀疑具有失常的区域。可以使用来自所鉴定区域的游离DNA测定组织类型的第一贡献率,且可以使用来自另一区域的游离DNA测定所述组织类型的第二贡献率。贡献率之间的显著分离表明所述组织类型是展现序列不均衡的组织类型,例如经由拷贝数失常所鉴定的序列不均衡或仅仅是针对已鉴定区域所测试的序列不均衡。I.通过甲基化解卷积所知的DNA混合物组成不同组织类型的基因组位点可以具有不同的甲基化水平。这些差异可以用于测定各种组织类型对混合物中的DNA的贡献率。因此,DNA混合物的组成可以通过组织特异性甲基化模式分析来测定。下述实例论述了甲基化密度,但可以使用其它甲基化水平。A.单一基因组位点甲基化解卷积原理可以使用单一甲基化基因组位点(甲基化标记)说明以测定生物体的DNA混合物组成。假定组织A的基因组位点完全甲基化,即甲基化密度(MD)为100%,且组织B完全未甲基化,即MD为0%。在此实例中,甲基化密度是指发生甲基化的具有CpG二核苷酸背景的胞嘧啶残基在所关注区域中的百分比。如果DNA混合物C是由组织A和组织B组成且DNA混合物C的总体甲基化密度是60%,那么我们根据下式可以推断组织A和B对DNA混合物C的贡献比例:MDC=MDA×a+MDB×b,其中MDA、MDB、MDC分别表示组织A、组织B和DNA混合物C的MD;且a和b是组织A和B对DNA混合物C的贡献比例。在此特定实例中,假定组织A和B是DNA混合物中仅有的两种成分。因此,a+b=100%。从而计算出组织A和B对DNA混合物的贡献分别是60%和40%。组织A和组织B中的甲基化密度可以获自生物体样品或获自相同类型的其它生物体的样品(例如潜在属于相同亚群的其它人)。如果使用来自其它生物体的样品,那么可以使用组织A样品的甲基化密度的统计分析(例如平均值、中值、几何平均值)获得甲基化密度MDA,且类似地获得MDB。可以选择个体间变异最小的基因组位点,例如小于特定的绝对变异量或处于所测试的基因组位点的最低部分内。举例来说,对于最低部分来说,实施例可以仅选择一组所测试基因组位点间具有最低10%变异的基因组位点。其它生物体可以取自健康个人,以及具有特定生理学的那些人(例如孕妇,或年龄不同的人或特定性别的人),其可以对应于包含所测试的当前生物体的特定亚群。亚群中的其它生物体也可以患有其它病理性病状(例如患有肝炎或糖尿病的患者等)。此亚群的不同组织可以具有改变的组织特异性甲基化模式。除使用正常组织的甲基化模式之外,可以使用组织在此类疾病条件下的甲基化模式进行解卷积分析。当测试来自患有那些病状的此亚群的生物体时,此解卷积分析可以更准确。举例来说,肝硬化肝脏或纤维化肾脏可以分别具有不同于正常肝脏和正常肾脏的甲基化模式。因此,如果针对其它疾病筛选肝硬化患者,那么可以更准确地是包括肝硬化肝脏作为候选组织之一(对血浆DNA有贡献之DNA),以及其它组织类型的健康组织。B.多个基因组位点当存在更多的潜在候选组织时,可以利用更多的基因组位点(例如10个或更多)测定DNA混合物的组成。DNA混合物的组成比例的估算准确性依赖于多种因素,包括基因组位点数目、基因组位点(也称为“位点”)相对于特定组织的特异性,以及测定参考组织特异性水平所利用的不同候选组织间和不同个体间的位点变异率。位点相对于组织的特异性是指特定组织与其它组织类型之间的基因组位点甲基化密度差异。其甲基化密度之间的差异越大,位点相对于特定组织的特异性就越强。举例来说,如果肝脏中的位点完全甲基化(甲基化密度=100%)而所有其它组织中的位点完全未甲基化(甲基化密度=0%),那么此位点对于肝脏来说具有高度特异性。然而,不同组织间的位点变异率可以通过例如(但不限于)不同类型组织中的位点甲基化密度的范围或标准差来反映。较大范围或较高标准差允许在数学上更精确且准确地测定不同器官对DNA混合物的相对贡献。这些因素对于估算候选组织对DNA混合物的贡献比例的准确性的影响说明于本申请的后续章节中。在此,我们利用数学方程式说明不同器官对DNA混合物的贡献比例的推导。DNA混合物中不同位点的甲基化密度与不同组织中相应位点的甲基化密度之间的数学关系可以如下表示:其中表示DNA混合物中位点i的甲基化密度;pk表示组织k对DNA混合物的贡献比例;MDik表示组织k中位点i的甲基化密度。当数目位点与器官数目相同或大于器官数目时,可以确定个体pk值。组织特异性甲基化密度可以获自其它个体,且可以选择个体间变异最小的位点,如上文所提及。算法中可以包括额外的准则以改进准确性。举例来说,所有组织的总计贡献可以限制为100%,即∑kpk=100%。另外,可以要求所有器官的贡献是非负的:由于生物变异,因此所观察到的总体甲基化模式与根据组织甲基化所推知的甲基化模式可能不完全相同。在此情形中,需要数学分析来测定个别组织最可能的贡献比例。就此而言,DNA中所观察到的甲基化模式与从组织推知的甲基化模式之间的差异用W表示。其中O是DNA混合物中观察到的甲基化模式且Mk是个别组织k的甲基化模式。pk是组织k对DNA混合物的贡献比例。每种pk最可能的值各自可以通过最小化W来确定,W是观察到的甲基化模式与推知的甲基化模式之间的差异。此方程式可以使用数学算法求解,例如通过使用二次规划、线性/非线性回归、期望最大化(EM)算法、最大似然算法、最大后验概率估算和最小二乘法。C.甲基化解卷积方法如上文所述,可以分析生物样品,包括来自生物体的游离DNA分子混合物,以测定混合物的组成,尤其是不同组织类型的贡献。举例来说,可以测定来自肝脏的游离DNA分子的贡献百分率。生物样品的贡献百分率的这些测量结果可以用于对生物样品进行其它测量,例如鉴定肿瘤位置,如下文章节中所述。图1是一个流程图,其图解说明根据本发明的实施例分析游离DNA分子的DNA混合物以利用甲基化水平测定不同组织类型的贡献率的方法100。生物样品包括来自M种组织类型的游离DNA分子的混合物。生物样品可以是各种实例中的任一个,例如如在此所提及。组织类型的数目M大于二。在各种实施例中,M可以是3、7、10、20或更大,或其间的任何数字。方法100可以使用计算机系统进行。在步骤110,鉴定N个基因组位点用于分析。N个基因组位点可以具有不同属性,例如如章节II中更详细所述,其描述I型和II型基因组位点。举例来说,N个基因组位点可以仅包括I型或II型位点,或两者的组合。基因组位点可以基于一或多个其它样品的分析加以鉴定,例如基于从关于在不同个体中所测量的甲基化水平的数据库获得的数据。在一些实施例中,N个基因组位点中至少10个是II型且每一个在M个组织类型间具有至少0.15的甲基化水平变异系数。可以使用更严格的变异系数阈值,例如0.25。M个组织类型的至少10个基因组位点中每一个的最大与最小甲基化水平之间的差异也可以超过0.1。可以使用更严格的变异系数阈值,例如0.2。N个基因组位点也可以包括I型位点(例如至少10个)。可以测量一个样品或一组样品的基因组基因座的这些甲基化性质。这组样品可以是含有所测试的本发明生物体的生物体亚群,例如与本发明生物体具有共同的特定性状的亚群。这些其它样品可以称为参考组织,且可以使用来自不同样品的不同参考组织。在步骤120,获得M个组织类型中每一个的N个基因组位点的N种组织特异性甲基化水平。N大于或等于M,以便组织特异性甲基化水平可以用于解卷积以测定百分比分率。组织特异性甲基化水平可以形成尺寸N×M的矩阵A。矩阵A中的每一列可以对应于特定组织类型的甲基化模式,其中所述模式是N个基因组位点的甲基化水平。在各种实施例中,组织特异性甲基化模式可以从公用数据库或此前研究中检索。在本文实例中,嗜中性白细胞和B细胞的甲基化数据是从高通量基因表达数据库(GeneExpressionOmnibus)(Hodges等人,《分子细胞(MolCell)》2011;44:17-28)中下载。其它组织(海马体、肝脏、肺、胰脏、心房、结肠(包括其不同部分,例如乙状结肠、横结肠、升结肠、降结肠)、肾上腺、食道、小肠和CD4T细胞)的甲基化模式是从表观组学路线图计划(RoadMapEpigenomicsproject)(Ziller等人,《自然(Nature)》2013;500:477-81)中下载。白细胞层、胎盘、肿瘤和血浆数据中的甲基化模式得自公开报道(Lun等人,《临床化学(ClinChem.)》2013;59:1583-94;Chan等人,《美国国家科学院院刊(ProcNatlAcadSciUSA.)》2013;110:18761-8)。这些组织特异性甲基化模式可用于鉴定N个基因组位点以用于解卷积分析中。在步骤130,接收包括来自M种组织类型的游离DNA分子混合物的生物样品。生物样品可以通过多种方式获自患者生物体。获得此类样品的方式可以无创或有创的。无创得到的样品实例包括某些类型的体液(例如血浆或血清或尿液)或粪便。举例来说,血浆包括来自多种器官组织的游离DNA分子,且因此适用于通过一种样品分析多种器官。在步骤140,分析生物样品中的游离DNA分子以鉴定其在与生物体对应的参考基因组中的位置。举例来说,可以对游离DNA分子进行测序以获得序列读数,且可以将序列读数与参考基因组对应(比对)。如果生物体是人类,那么参考基因组是潜在地来自特定亚群的参考人类基因组。作为另一实例,可以使用不同探针分析游离DNA分子(例如在PCR或其它扩增之后),其中每个探针对应于不同基因组位点。在一些实施例中,游离DNA分子的分析可以通过接收对应于游离DNA分子的序列读数或其它实验数据且然后分析实验数据来进行。可以分析游离DNA分子的统计显著数目以便提供准确的解卷积用于测定M个组织类型的贡献率。在一些实施例中,分析至少1,000个游离DNA分子。在其它实施例中,可以分析至少10,000或50,000或100,000或500,000或1,000,000或5,000,000个游离DNA分子或更多。待分析的分子总数可以依赖于M和N以及期望的精确度(准确度)。在步骤150,使用第一组游离DNA分子测量N个基因组位点的N种混合甲基化水平,所述第一组游离DNA分子各自位于参考基因组的N个基因组位点中的任一个处。N个混合甲基化水平是指生物样品混合物中的甲基化水平。作为实例,如果混合物中的游离DNA分子位于N个基因组位点之一,那么位于所述位点的那个分子的甲基化指数可以包括于那个位点的总体甲基化密度中。N个混合甲基化水平可以形成长度N的甲基化向量b,其中b对应于可以据以测定组织类型贡献率的观察值。在一个实施例中,DNA混合物中的基因组位点的甲基化水平可以使用全基因组亚硫酸氢盐测序法来测定。在其它实施例中,基因组位点的甲基化水平可以使用甲基化微阵列分析进行测定,如IlluminaHumanMethylation450系统,或使用甲基化免疫沉淀(例如使用抗甲基胞嘧啶抗体)或用甲基化结合蛋白处理,随后进行微阵列分析或DNA测序,或使用甲基化敏感性限制酶处理,随后进行微阵列或DNA测序,或使用甲基化感知测序,例如使用单分子测序方法(例如纳米孔测序(Schreiber等人,《美国国家科学院院刊》2013;110:18910-18915)或PacificBiosciences的单分子实时分析(Flusberg等人,《自然方法(NatMethods)》2010;7:461-465))。组织特异性甲基化水平可以用相同方式测量。作为其它实例,可以利用靶向亚硫酸氢盐测序、甲基化特异性PCR、基于非亚硫酸氢盐的甲基化感知测序(例如通过单分子测序平台(Powers等人,有效且准确的大肠杆菌全基因组组合体和甲基化组特征(EfficientandaccuratewholegenomeassemblyandmethylomeprofilingofE.coli),《BMC基因组学(BMCGenomics)》2013;14:675)来分析血浆DNA的甲基化水平用于血浆DNA甲基化解卷积分析。因此,甲基化感知测序结果可以通过多种方式获得。在步骤160,测定组成向量的M值。每个M值对应于M个组织类型中的特定组织类型对DNA混合物的贡献率。已知NxM个组织特异性甲基化水平,可以对组成向量的M值求解以得到N个混合甲基化水平(例如甲基化向量b)。M个贡献率可以对应于向量x,向量x通过对Ax=b求解来测定。当N大于M时,解法可以涉及误差最小化,例如使用最小二乘法。在步骤170,利用组成向量测定M个组织类型中的每一个在混合物中的量。组成向量的M值可以直接视为M个组织类型的贡献率。在一些实施方案中,M值可以换算成百分率。误差项可以用于将M值转换成更高或更低的值。每一个组成向量值可以视为一个分量,且第一分量可以对应于第一组织类型。D.应用如上文所提及,贡献率可以用于进一步测量生物样品和其它测定,例如特定染色体区域是否具有序列不均衡或特定组织类型是否发生病变。图2绘示了一个示意图,其根据本发明的实施例绘示了DNA甲基化解卷积的若干潜在应用(例如使用血浆)。在图2中,在210对生物样品205进行全基因组亚硫酸氢盐测序。在230,血浆DNA组织映射是利用组织特异性甲基化概况220测定组织贡献百分率。实例组织特异性甲基化概况是以肝脏、血细胞、脂肪组织、肺、小肠和结肠绘示。贡献百分率可以如上文和别处所述测定,例如对Ax=b求解。适用于测定不同器官对血浆DNA的贡献的甲基化标记(基因组位点)清单可以通过比较不同组织(包括肝脏、肺、食道、心脏、胰脏、乙状结肠、小肠、脂肪组织、肾上腺、结肠、T细胞、B细胞、嗜中性白细胞、脑和胎盘)甲基化特征(图2)加以鉴定。在不同实例中,从得自贝勒医学院(BaylorCollegeofMedicine)的人类表观基因组图谱(HumanEpigenomeAtlas)(www.genboree.org/epigenomeatlas/index.rhtml)中检索肝脏、肺、食道、心脏、胰脏、结肠、小肠、脂肪组织、肾上腺、脑和T细胞的全基因组亚硫酸氢盐测序数据。B细胞和嗜中性白细胞的亚硫酸氢盐测序数据获自Hodges等人的出版物(Hodges等人;定向DNA甲基化变化和复杂中间状态伴随成人造血区室中的谱系特异性(DirectionalDNAmethylationchangesandcomplexintermediatestatesaccompanylineagespecificityintheadulthematopoieticcompartment),《分子细胞》2011;44:17-28)。胎盘的亚硫酸氢盐测序数据得自Lun等人(Lun等人,《临床化学(ClinChem)》2013;59:1583-94)。在其它实施例中,可以从使用微阵列分析(例如使用IlluminaInfinium人类甲基化450BeadChip阵列)所产生的数据集中鉴定出标记。II.甲基化标记的选择我们在上文中已经描述利用甲基化分析测定DNA混合物组成的原理。具体地说,可以利用甲基化分析来测定不同器官(或组织)对血浆DNA的贡献百分率。在这个章节中,我们进一步描述选择甲基化标记的方法和这项技术的临床应用。通过甲基化分析测定DNA混合物组成的结果受到DNA混合物组成解卷积所用的甲基化标记的影响。因此,适当基因组甲基化标记的选择对于DNA混合物组成的准确测定来说可为重要的。A.甲基化标记解卷积准则选择标记时,可以考虑以下三种属性。(i)期望甲基化标记在不同个体间的相同组织类型中所测量的甲基化水平方面具有较低变异率。由于DNA混合物组成的测定依赖于对组织特异性甲基化模式的认知,因此不同个体间的相同组织类型中的较低甲基化水平变异率适用于准确鉴定DNA混合物中的组织特异性模式。在组织特异性甲基化水平获自其它生物体样品(例如获自数据库)的实施例中,较低变异率意味着其它样品的甲基化水平类似于所测试的当前生物体的组织特异性甲基化水平。(ii)期望甲基化标记在不同组织间的甲基化水平方面具有高度变异率。对于特定标记来说,不同组织间的甲基化水平的较高差异可以更精确地测定不同组织对DNA混合物的贡献。具体地说,精确度的改善可以通过使用具有属性(ii)的一组标记和具有属性(iii)的另一组标记来获得。(iii)期望甲基化标记在特定组织中的甲基化水平与大部分或全部其它组织中的甲基化水平相差悬殊。与上述第(ii)点相比,标记在大部分组织的甲基化水平方面可以具有较低变异率,但其在一种特定组织中的甲基化水平不同于大部分其它组织。此标记特别适用于测定具有不同于其它组织的甲基化水平的组织的贡献。B.实例标记选择原理说明于表1中的以下假想实例中。表1.不同组织中6种假想甲基化标记的甲基化密度。在这个假想实例中,标记2在来自三位个体的肝脏的甲基化密度方面的变异率低于标记1。因此,标记2优于标记1之处是作为测定肝脏对DNA混合物的贡献的标志。与标记4相比,标记3在不同组织类型间的甲基化密度方面具有较高变异率。根据上文所论述的数学关系,不同组织的所估计贡献的相同变化水平使得标记3的DNA混合物的推知甲基化密度的变化大于标记4。因此,使用标记3可以更精确地估计每种组织的贡献。标记5在肝脏、心脏和肺间的甲基化密度的变异率较低。其甲基化密度为10%到14%不等。然而,结肠的甲基化密度是80%。此标记特别适用于测定结肠对DNA混合物的贡献。类似地,对于标记6来说,心脏的甲基化低于其它组织。因此,可以通过标记6来准确地测定心脏的贡献。因此,标记5与标记6的组合能够准确地测定结肠和心脏的贡献。标记2和标记3的增添然后足以推断出四种器官(包括肝脏、心脏、肺和结肠)中的每一种的贡献。C.不同类型的标记甲基化标记可能不一定需要具有所有上述三种属性。I型甲基化标记典型地具有上述属性(iii)。多个此类标记还可以具有属性(i)。另一方面,II型甲基化标记典型地具有上述属性(ii)。多个此类标记还可以具有属性(i)。还有可能的是,特定标记可以具有所有三种属性。在一些实施例中,标记广泛分成两个类型(I型和II型)。I型标记具有组织特异性。这些标记在一组特定的一或多种组织中的甲基化水平不同于大部分其它组织。举例来说,相较于所有其它组织的甲基化水平,特定组织的甲基化水平可以是显著的。在另一个实例中,两种组织(例如组织A和组织B)具有相似的甲基化水平,但组织A和组织B的甲基化水平显著不同于其余组织的甲基化水平。II型标记具有高度的组织间甲基化变异率。这些标记的甲基化水平在不同组织间是高度可变的。此类别中的单个标记可能不足以测定特定组织对DNA混合物的贡献。然而,II型标记的组合,或II型标记与一或多种I型标记的组合可以共同用于推断个别组织的贡献。依据上述定义,特定标记可以是单独的I型标记、单独的II型标记,或同时是I型和II型标记。1.I型标记在一个实施例中,可以通过对标记的甲基化密度与此特定标记在所有候选组织中的甲基化密度的平均值和标准差(SD)进行比较来鉴定I型标记。在一个实施方案中,如果一种标记在一种组织中的甲基化密度与所有组织的平均值相差3个标准差(SD),那么鉴定标记。研究获自上述来源的14种组织的甲基化概况以选择标记。在一项分析中,使用上述准则鉴定总共1,013个I型标记(附录A的表S1中标识为I型的标记)。在其它实施例中,可以使用特定组织与平均甲基化密度之间的其它截止值,例如1.5SD、2SD、2.5SD、3.5SD和4SD。在又另一个实施例中,可以通过对特定组织的甲基化密度与所有组织的中值甲基化密度进行比较来鉴定I型标记。在其它实施例中,当超过一种组织(例如两种、三种、四种或五种组织)展示的甲基化密度显著不同于所有候选组织的平均甲基化密度时,可以获得I型标记。在一个实施方案中,可以利用所有候选组织的甲基化密度的平均值和SD来计算截止甲基化密度。出于说明的目的,截止(阈值水平)可以定义为比平均甲基化密度高或低3SD。因此,当指定数目个组织的甲基化密度比平均甲基化密度高大于3SD或比组织的平均甲基化密度低大于3SD时,可以选择标记。2.II型标记为了鉴定II型标记,计算所有14种候选组织间的甲基化密度平均值和SD且SD与平均值的比率表示为变异系数(CV)。在此说明性实例中,我们使用>0.25的CV截止值鉴定合格的II型标记,以及组织群组的最大与最小甲基化密度之间的差异超过0.2。使用这些准则鉴定出5820个II型标记(附录A的表S1中标识为II型的标记)。CV截止值的其它实例包括0.15、0.2、0.3和0.4。最大与最小甲基化密度之间差异的截止值的其它实例包括0.1、0.15、0.25、0.3、0.35、0.4、0.45和0.5。在其它实施例中,相同组织类型的多个样品间的平均值可以用于测量不同组织间的甲基化水平的变异率。举例来说,可以对10种样品的相同基因组位点的10种甲基化水平取平均值以获得基因组位点的单一甲基化水平。可以执行类似方法来测定其它组织类型的基因组位点的平均甲基化水平。然后可以利用组织类型间的平均值确定基因组位点在组织类型间是否具有显著的变异。除平均值之外,可以使用其它统计值,例如中值或几何平均值。此类统计值可以用于鉴定I型和/或II型标记。相同组织类型的不同样品(例如来自不同个体)可以用于测定甲基化水平在不同样品间的变异。因此,如果相同组织类型存在多个样品,那么实施例可以进一步测量特定标记在相同组织类型的此类样品间的变异。样品间变异低的标记是比高变异标记更可靠的标记。实施例还涉及表S1中的标记以及使用标记的任何组合,例如使用表S1中的任何10个或更多个I型或II型标记,以及来自每个表的10个或更多个的任何组合。举例来说,实施例涉及使用来自表S1的50(或100、250、500或1,000)个I型标记和50(或100、250、500、1,000、2,000或5,000)个II型标记。D.不同类别的标记基因组基因座的“类别”(甲基化标记)对应于相同组织类型的基因座的甲基化水平在不同个体间的的具体变异。不同类别在个体间、在特定组织类型间可以具有不同的变异范围。在所测试的个体间,第一类甲基化标记的甲基化水平可能具有10%或更低的差异。在所测试的个体间,第二类甲基化标记的甲基化水平可能具有超过10%的差异。使用个体间变异较低的甲基化标记(第一类标记)潜在地提高了测定特定器官对DNA混合物的贡献的准确度。E.潜在甲基化标记的鉴定在一些实施例中,按以下方式鉴定潜在的甲基化标记。然后可以对此类潜在的甲基化标记执行上述准则以鉴定I型和II型标记。在其它实施例中,不需要I型或II型的鉴定。并且,其它实施例可以利用其它技术鉴定潜在的甲基化标记。在一些实施例中,常染色体上的所有CpG岛(CGI)和CpG岸均视为潜在的甲基化标记。不使用性染色体上的CGI和CpG岸,以便将源数据中的与性相关染色体剂量差异有关的甲基化水平的变异减到最少。CGI是从加利福尼亚大学圣克鲁兹(UniversityofCalifornia,SantaCruz)(UCSC)数据库下载(genome.ucsc.edu/,人类基因组存在27,048个CpG岛)(Kent等人,UCSC的人类基因组浏览器(ThehumangenomebrowseratUCSC),《基因组研究(GenomeRes.)》2002;12(6):996-1006)且CpG岸定义为CpG岛的2kb侧接窗(Irizarry等人,人类结肠癌甲基化组在保守性组织特异性CpG岛岸展示出类似的低甲基化和高甲基化(Thehumancoloncancermethylomeshowssimilarhypo-andhypermethylationatconservedtissue-specificCpGislandshores),《自然遗传学(NatGenet)》2009;41(2):178-186)。然后,将CpG岛和岸细分成不重叠的500bp单元且每个单元视为潜在的甲基化标记。比较14种组织类型之间的所有潜在基因座的甲基化密度(即,500bp单元内的甲基化CpG百分率)。如此前所报道(Lun等人,《临床化学》2013;59:1583-94),发现胎盘出现全面低甲基化(与其余组织相比)。因此,在标记鉴定期不包括胎盘的甲基化特征。利用其余13种组织类型的甲基化特征,鉴定出两种类型的甲基化标记。举例来说,I型标记可以指一种组织中的甲基化密度比13种组织类型的平均水平低或高3个SD的任何基因组位点。当(A)最高甲基化组织的甲基化密度比最低甲基化组织的甲基化密度高至少20%时;以及(B)所有13种组织类型的甲基化密度的SD当除以群组的平均甲基化密度(即变异系数)时是至少0.25时,II型标记可以视为高度可变的。最后,为了减少潜在冗余标记的数目,在侧接一个CpG岛的两个CpG岸的一个邻接嵌段中可以选择仅一个标记。F.基于应用的选择针对特定应用所选择的甲基化标记组可以依据所期望应用的参数改变。举例来说,为了确定基因组失常(例如拷贝数失常(CNA))的起源,散布于整个基因组中的大量标记是有利的。作为另一实例,在DNA从特定组织释放到血浆中特别显著的应用中,可以优先选择在此组织类型中甲基化有差异的数目大于标记组中的其它标记的甲基化标记(例如I型标记)。解卷积分析中甲基化标记的数目和选择可以根据预定用途而改变。如果肝脏的贡献率尤其受关注(例如在已接受肝脏移植的患者中),那么可以在解卷积分析中使用更多的I型肝脏特异性标记以增加定量所移植肝脏对血浆DNA的贡献的精确度。III.组成准确度如上文所述,实施例可以鉴定组织对血浆DNA的贡献。在不同实例中,对血浆DNA进行全基因组亚硫酸氢盐测序且参照不同组织的甲基化特征进行分析。作为实例,利用二次规划对血浆DNA测序数据进行解卷积,得到不同组织的贡献比例。对孕妇、患有肝细胞癌、肺癌和结肠直肠癌的患者以及骨髓和肝脏移植后的个体的实施例进行测试。在大部分个体中,白血细胞是循环DNA池的主要贡献者。孕妇中的胎盘贡献与贡献比例相关,如通过胎儿特异性遗传标记所揭露。移植接受者血浆的移植物来源性贡献与利用供体特异性遗传标记所测定的那些贡献相关。患有肝细胞癌、肺癌或结肠直肠癌的患者表明患有肿瘤的器官对血浆DNA的贡献提高。肝细胞癌患者的肝脏贡献还与利用肿瘤相关拷贝数失常所得到的测量结果相关。在血浆展现拷贝数失常的癌症患者和孕妇中,甲基化解卷积查明了造成失常的组织类型。在经诊断在怀孕期间患有滤泡性淋巴瘤的孕妇中,甲基化解卷积指出B细胞对血浆DNA池的贡献大体上提高且局部B细胞(而非胎盘)为血浆中所观察到的拷贝数失常的起源。相应地,实施例可以充当有效的工具,以便基于不同组织对血浆的扰动贡献比例的鉴定来评估广泛范围的生理学和病理学病状。A.不同类型的血细胞的贡献作为甲基化解卷积的一个实例,我们测定不同组织和细胞类型对循环DNA的贡献。从两位罹患全身性红斑狼疮(systemiclupuserythematosus,SLE)的患者中收集两个血液样品。收集之后,将静脉血液样品以1,500g离心10分钟。离心之后,分离血细胞和血浆。然后从血细胞中提取DNA。DNA用亚硫酸氢盐转化且在HiSeq2000测序仪中使用流动池中的一个泳道进行测序。使用细胞型特异性甲基化模式分析对两个血细胞样品进行分析。作为血细胞DNA的潜在候选物,包括嗜中性白细胞、淋巴细胞、食道、结肠、胰脏、肝脏、肺、心脏、肾上腺和海马体的甲基化模式。选择609个甲基化标记进行分析。还传送两位个体的全血样品用于细胞计数以测定嗜中性白细胞和淋巴细胞在血细胞中的组成分率。表2.通过解卷积模式分析和细胞计数得知的血液组织贡献在甲基化模式分析中,嗜中性白细胞和淋巴细胞是作为组成血细胞DNA的主要组分进行测定。根据细胞计数分析,嗜中性白细胞和淋巴细胞的相对贡献比例类似于其在血液样品中的相对丰度。B.孕妇利用孕妇血浆DNA的甲基化分析对不同组织(包括肝脏、肺、胰脏、结肠、海马体、小肠、血细胞、心脏、肾上腺、食道和胎盘)的贡献进行分析。由于胎盘基因型通常与胎儿基因型相同,但与孕妇基因型不同,因此胎盘对母体血浆的精确贡献可以通过对样品中的胎儿特异性等位基因的数目进行计数来准确测定。1.组成和与胎儿DNA百分率的相关性对15位孕妇的血浆DNA进行全基因组亚硫酸氢盐测序,第一、第二和第三个三月期各选五位。进行甲基化解卷积且推导不同组织的贡献百分率。利用二次规划分析,基于表S1中所有I型和II型标记的甲基化水平(如甲基化密度)来测定不同器官的贡献。图3A根据本发明的实施例绘示了15位孕妇的不同器官对血浆DNA的贡献百分率图300。每一条形图对应于一个样品的结果。不同颜色表示不同器官对血浆的贡献。这些结果表明白血细胞(即嗜中性白细胞和淋巴细胞)是血浆DNA池的最重要贡献者。这个观察结果符合此前在骨髓移植后所得的那些观察结果(LuiYY等人,《临床化学》2002;48:421-7)。图4绘示了根据本发明的实施例,利用孕妇间的血浆DNA组织映射分析所测定的贡献百分率表400。这些结果还表明胎盘是孕妇中的血浆DNA的另一个关键贡献者,其浓度分率为9.9%到38.4%。我们还利用孕妇所没有的从父亲继承的胎儿单核苷酸多态性(SNP)等位基因测量了胎盘贡献。为了分析胎儿特异性SNP等位基因,通过分析绒毛样品或胎盘来确定胎儿基因型。通过分析血细胞来确定孕妇基因型。基于SNP的结果表明甲基化解卷积结果得到独立的校验。图3B根据本发明的实施例绘示了图350,其是根据血浆DNA甲基化解卷积所推知的胎盘所贡献的血浆DNA分率与使用胎儿特异性SNP等位基因所推知的胎儿DNA分率之间的相关性。图350表明通过甲基化解卷积所测定的胎盘贡献与利用SNP所测量的胎儿DNA分率强烈相关(r=0.99,p<0.001,皮尔逊相关系数(Pearsoncorrelation))。相应地,观察到两个参数的值之间存在良好的正相关,表明血浆DNA甲基化解卷积准确地测定了胎盘对母体血浆样品的贡献。图5根据本发明的实施例、依据血浆DNA组织映射和基于胎儿特异性SNP等位基因的胎儿DNA分率绘示了除胎盘之外的器官的贡献百分率图。X轴表示通过基于SNP的分析所估计的胎儿DNA分率且Y轴表示通过血浆组织DNA映射分析所推导的贡献百分率。嗜中性白细胞对血浆DNA的贡献展示逆相关。这可能是由于以下事实:嗜中性白细胞是血浆DNA池的主要贡献者且因此,随着胎盘贡献增加,嗜中性白细胞的相对贡献必然减少。其余组织的甲基化解卷积结果表明与胎儿DNA分率不相关。图6根据本发明的实施例绘示了得自非怀孕健康对照个体间的血浆DNA组织映射分析的贡献百分率表600。当对非怀孕健康对照的血浆应用方法时,胎盘贡献就大部分样品来说是缺乏的(中值:0%;四分位区间:0%到0.3%)。2.所选标记相对于随机标记的比较利用选择标记相对于随机标记来测试贡献百分率准确度。不同标记组进行不同的组成计算。一组是基于上文所提及的准则选择,且另一组是随机组。结果表明为了获得准确结果,慎重选用甲基化标记(基因组基因座)是重要的。此分析招募十一位孕妇和四位健康非怀孕个体。其血浆DNA经亚硫酸氢盐转化且使用IlluminaHiSeq2000测序仪测序。每个血浆样品使用测序流动池中的一个泳道测序。然后利用生物信息学程序Methy-Pipe(JiangP.,《科学公共图书馆综合卷(PLoSOne)》2014;9:e100360)分析所测序的读数。这个程序可以将亚硫酸氢盐转化的序列读数与参考基因组进行比对且测定每个测序片段上的每个CpG位点的甲基化状况。因此,可以利用各自与参考基因组中的至少一个基因组位点比对的序列读数测量混合甲基化水平。第一组标记对于利用血浆DNA鉴定不同组织具有高度特异性。对于每种组织类型来说,选择甲基化密度与其它组织相比具有最大差异的标记。标记是利用含有至少一个CpG二核苷酸的基因组区域测定。在此实例中,CpG岛(CGI)用作潜在标记,其在DNA的特定片段中具有高频率的CpG位点。在此特定实例中,CGI是从加利福尼亚大学圣克鲁兹(UCSC)数据库下载:(genome.ucsc.edu)。我们从人类基因组获得总计27,048个CpG岛。CpG岛d中值尺寸是565bp(范围:200bp到45kb)。90%岛小于1.5kb。对于每个甲基化标记来说,测定所关注组织与其它组织之间的甲基化密度差异。然后以所有其它组织的标准差(SD)数目表示所述差异。对于所关注的组织来说,根据甲基化密度的这种差异来对所有标记评级。选择最大差异高于(10个标记)和低于(10个标记)其它组织的平均甲基化密度的20个标记。标记数目的其它实例包括5、15、20、30、40、50、100和200。另外,还选择所有不同组织间具有高度变异率的标记。在此实例中,选择甲基化密度最高与最低的组织之间差异>50%的标记。差异值的其它实例包括20%、30%、40%、60%、70%和80%。另外,不同组织间的甲基化密度变异率也基于平均值和SD计算。在此实例中,如果SD值是平均值的大于两倍,那么也选择标记。截止值的其它实例可以包括1、1.5、2.5和3的标准差。基于这些选择准则,选择344个甲基化标记作为第一组。从上文所论述的27,048个CGI中随机选择341个标记作为第二组。所有CGI首先从1到27,048编号。然后,通过计算机产生随机数目(1到27,048之间)用于选择标记。然后重复这个过程,直到选择总共341个标记。如果已经使用所产生的随机数目,那么会产生另一个数目。这组标记预期在鉴定组织特异性甲基化模式时具有低得多的特异性。因此,预期血浆DNA组成的测定准确度降低。图7根据本发明的实施例绘示了使用第一组标记(具有高度器官特异性)估算的11位孕妇和4位非怀孕健康个体的不同器官对血浆DNA的贡献率表700。通过对胎儿特异性等位基因进行计数来测定胎儿DNA分率且展示于底行中。在四位非怀孕对照个体的每一个中,胎盘对血浆的贡献经测定接近于0%。这表明这种方法的特异性。图8根据本发明的实施例绘示了使用第二组标记(器官特异性较低)估算的11位孕妇和4位非怀孕健康个体的不同器官对血浆DNA的贡献表800。通过对胎儿特异性等位基因计数所测定的胎儿DNA分率展示于底行中。利用这些较少特异性标记,观察到胎盘的贡献百分率相对不一致,且在四位非怀孕对照个体中观察到来自胎盘的相当大贡献。这表明标记的组织特异性在这种方法中是重要的。图9A是图900,其绘示了所估算的胎儿DNA分率(胎盘的贡献)与通过对母体血浆样品中的胎儿特异性等位基因计数所测定的胎儿DNA分率之间的相关性。通过使用第一组甲基化标记,两种技术产生的结果具有良好相关性。然而,通过使用第二组甲基化标记,使用甲基化分析的估算表明显著偏离使用胎儿特异性等位基因计数所测定的真实值。图9B是图950,其绘示了利用甲基化标记的估算与通过胎儿特异性等位基因计数所测定的胎儿DNA分率之间的绝对差。通过使用第一组标记和第二组标记,使用甲基化分析估算的中值误差分别是4%和8%。C.癌症患者实施例还能够用于测定癌症患者血浆中的癌源DNA的量。在这个实例中,从10位罹患肝细胞癌(HCC)的患者中收集静脉血液样品。利用如上文所述的组织特异性甲基化模式分析来测定不同器官(包括肝脏、肺、结肠、小肠、胰脏、食道、肾上腺、心脏、脑和血细胞)的贡献百分率。另外,还利用亚硫酸氢盐测序来分析肿瘤组织,以鉴定肿瘤特异性甲基化模式。所有不同组织的结果取平均值以确定代表性肿瘤组织模式。利用这些肿瘤特异性甲基化标记,还测定肿瘤对血浆DNA的贡献。此分析中使用总共828个器官特异性标记。作为对照,分析中还包括无癌症的四位健康对照个体。对于各种情况来说,根据血浆的总甲基化水平来测定癌症患者中的肿瘤组织对血浆DNA的实际贡献。已经表明肿瘤组织的甲基化通常低于非肿瘤组织(Feinberg等人,《自然(Nature)》1983;301:89-92和Chan等人,《美国国家科学院院刊》2013;110:18761-8)。非恶性组织的全基因组甲基化水平是约70%,而肿瘤组织的全基因组甲基化水平是约45%。因此,肿瘤对血浆DNA的贡献可以利用下式估算:f×45%+(1-f)×75%=MDP其中MDP是血浆样品中所测量的全基因组甲基化水平且f是血浆中肿瘤源DNA的浓度分率。这种估算肿瘤源DNA分率的方法已经表明与基于检测染色体失常的方法非常相关(Chan等人,《美国国家科学院院刊》2013;110:18761-8)。图10根据本发明的实施例、基于器官特异性甲基化模式分析绘示了癌症患者和健康患者的不同组织对血浆DNA的贡献表1000。在未患癌症的四位健康对照个体的每一个中,肿瘤组织的贡献经测定为0%。这表明甲基化模式分析具有特异性。图11A是图1100,其根据本发明的实施例绘示了通过器官特异性甲基化模式分析所测定和根据全基因组甲基化水平所测定的肿瘤DNA分率值。图1100绘示了在10位HCC患者中,通过器官特异性甲基化模式分析所测定的肿瘤DNA分率与通过全基因组甲基化水平分析所测定的肿瘤DNA分率非常相关。我们还通过研究异型接合性有损失的基因组区域测量了血浆中的HCC肿瘤DNA的浓度分率,这是一种此前我们已经称为全基因组汇总等位基因损失(genome-wideaggregatedallelicloss,GAAL)的技术(ChanKCA等人(2013)《临床化学》59(1):211-224)。图11B是图1150,其绘示了肝脏所贡献的血浆DNA分率(基于血浆DNA组织映射分析)与通过GAAL分析所测定的肿瘤源血浆DNA分率之间的相关性。图1150表明通过甲基化解卷积所推知的血浆肝源DNA贡献与通过GAAL所测量的肿瘤DNA浓度之间存在良好相关性(r=0.63,p=0.015,皮尔逊相关系数)。在另一个实施例中,全基因组汇总等位基因损失(GAAL)分析可以按如下方式进行。HCC病例的肿瘤样品可以使用Affymetrix全基因组人类SNP阵列6.0系统进行分析。展现异型接合性损失(lossofheterozygosity,LOH)的区域可以如此前所述进行鉴定(Chan等人,《临床化学》2013;59:211-24)。血浆中的肿瘤源DNA浓度分率可以使用以下方程式,通过按全基因组方式分析血浆测序数据中展现LOH的SNP的等位基因计数来测定:其中Nnon-del表示肿瘤组织中携带非缺失等位基因的测序读数的数目,且Ndel表示肿瘤组织中携带缺失等位基因的测序读数的数目。图12A是图1200,其绘示了患者HCC10在不同时间的血浆中的估算肿瘤源DNA。手术之前取样(Pre-Tx)且在患者的手术切除之后第3天和第3个月取样。这位患者在肿瘤切除之后第2年临床缓解。在手术切除肿瘤之后第3天和第3个月,血浆中检测不到肿瘤特异性甲基化模式。这个发现与手术之后第2年缺乏任何可检测癌症的发现相容。图12B是图1250,其绘示了患者HCC9血浆中的估算肿瘤源DNA。治疗之前取样(Pre-Tx)且在患者的手术切除之后第3天和第2个月取样。随后诊断出这位患者的剩余未切除肝脏中在第3个月出现多病灶肿瘤沉积物(此前在手术时未知)且注意到在手术之后的第4个月出现多个肺转移。患者在手术之后第8个月死于转移性疾病。使用组织特异性甲基化模式分析,估算出肿瘤组织对总血浆DNA的贡献在手术之后的第3天和第2个月为8%和12%。D.器官移植和解卷积定量器官对血浆DNA的贡献可以有效地应用于监视接受器官移植的患者。已经表明,移植器官所释放的DNA的含量在与移植器官损伤有关的情形下,例如在组织排斥反应中会增加(DeVlaminck等人,《科学·转化医学(SciTranslMed)》2014;6:241ra77)。然而,现有方法仅基于检测供者与接受者之间不同的多态性标记,例如存在于供者中、但不存在于接受者中的SNP等位基因(DeVlaminck等人,循环中的游离DNA能够实现心脏移植排斥反应的无创诊断(Circulatingcell-freeDNAenablesnoninvasivediagnosisofhearttransplantrejection),《科学·转化医学》2014;6:241ra77),或性别错配移植情况中的染色体Y序列(GarcíaMoreira等人,游离DNA作为肾移植中的无创急性排斥反应标记(Cell-freeDNAasanoninvasiveacuterejectionmarkerinrenaltransplantation),《临床化学》2009;55:1958-66)。为了分析多态性标记,器官供者与接受者的组织均需要进行基因分型。供者和接受者组织的基因分型增添了分析的额外成本且器官供者的组织在实际上可能无法利用。并且,染色体X和Y上的序列仅适用于供者与接受者性别不同的情形中。因此,相较于一些此前技术和更适用的其它此前技术,甲基化解卷积技术可以是时间较少而成本密集的。1.分率相关性这个章节表明测定供者器官所贡献的血浆DNA的比例的准确度,如利用血浆DNA甲基化解卷积分析所测定。在此方法中,不需要对供者和接受者的组织进行基因分型。已经接受移植的个体为验证血浆DNA组织映射方法提供了宝贵的机会。通过使用存在于器官供者中而不存在于移植接受者中的SNP等位基因,可以如此前所述测量移植器官在血浆中的浓度分率(ZhengYW等人,2012)。然后可以对这个结果与利用甲基化解卷积所推导的结果进行比较。图13是表1300,其根据本发明的实施例绘示器官移植患者间的血浆DNA组织映射分析。我们对4位肝脏移植接受者和3位骨髓移植接受者进行血浆DNA组织映射。对于每种情况来说,获得供者和接受者的组织且利用大规模平行测序进行基因分型。鉴定出供者特异性SNP等位基因且用于计算供者器官所贡献的血浆DNA的分率。根据肝脏移植接受者间的肝脏贡献,对使用供者特异性SNP等位基因所估算的供者DNA分率进行比较,同时根据白细胞贡献(即嗜中性白细胞加淋巴细胞),对骨髓移植接受者间的那些贡献进行比较。然后进行血浆甲基化解卷积以测定肝脏和血细胞分别在肝脏移植情况和骨髓移植情况中的贡献。图14是图1400,其绘示了根据血浆DNA组织映射所推知的移植物所贡献的血浆DNA分率与使用供者特异性SNP等位基因所测定的供者DNA分率之间的相关性。三角形表示肝脏移植接受者的结果且圆点表示骨髓移植接受者的结果。图1400绘示了甲基化解卷积与基于SNP的结果之间的强烈相关性(r=0.99,p<0.001,皮尔逊相关系数)。2.不同标记类型的比较对甲基化解卷积分析中的I型和II型标记的相对贡献进行比较。为了对其贡献进行充分的比较,首先随机选择1013个II型标记,使得连续分析中所用的I型标记与II型标记的数目相同。1013个I型标记和1013个II型标记形成池。使用随机选择的不同数目个甲基化标记进行甲基化解卷积,以测定移植器官(即肝脏移植接受者的肝脏和骨髓移植接受者的血细胞)的贡献。标记随机选择之后,基于实际测序数据进行解卷积分析。在每种分析中,使用相同数目个I型和II型标记。然而,在解卷积分析的不同组中,标记总数是变化的,以便确定标记数目对甲基化解卷积分析准确度的影响。在每种分析中,对通过甲基化解卷积所得到的移植器官对血浆DNA的贡献百分率与供者特异性SNP等位基因所产生的值之间的差异作图。图15A是图1500,其绘示了一种分析,所述分析对使用503个I型、503个II型以及两种类型(各503个)标记进行甲基化解卷积的准确性进行了比较。针对已经接受肝脏移植(LTP1到LTP5)的患者且针对已经接受骨髓移植(BMT1到BMT3)的患者,绘示通过甲基化解卷积所得到的移植器官对血浆DNA的贡献百分率与供者特异性SNP等位基因所产生的值之间的差异。针对每一患者,在左侧、在中间和在右侧用方框分别绘示使用单独I型标记、单独II型标记和两种类型标记的甲基化解卷积结果。使用单独I型标记的分析的偏差大于使用单独II型标记,或这两种类型的标记。另一方面,使用单独II型标记的结果与使用这两种类型标记的结果之间未观察到显著差异。图15B是图1550,其绘示了一种分析,所述分析对使用251个I型、251个II型以及两种类型(各251个)标记进行甲基化解卷积的准确性进行了比较。针对已经接受肝脏移植(LTP1到LTP5)的患者且针对已经接受骨髓移植(BMT1到BMT3)的患者,绘示通过甲基化解卷积所得到的移植器官对血浆DNA的贡献百分率与供者特异性SNP等位基因所产生的值之间的差异。针对每一患者,在左侧、在中间和在右侧用方框分别绘示使用单独I型标记、单独II型标记和两种类型标记的甲基化解卷积结果。使用单独I型标记的分析的偏差大于使用单独II型标记,或这两种类型的标记。另一方面,使用单独II型标记的结果与使用这两种类型标记的结果之间未观察到显著差异。图16A是图1600,其绘示了一种分析,所述分析对使用123个I型、123个II型以及两种类型(各123个)标记进行甲基化解卷积的准确性进行了比较。针对已经接受肝脏移植(LTP1到LTP5)的患者且针对已经接受骨髓移植(BMT1到BMT3)的患者,绘示通过甲基化解卷积所得到的移植器官对血浆DNA的贡献百分率与供者特异性SNP等位基因所产生的值之间的差异。针对每一患者,在左侧、在中间和在右侧用方框分别绘示使用单独I型标记、单独II型标记和两种类型标记的甲基化解卷积结果。使用单独I型标记的分析的偏差大于使用单独II型标记,或这两种类型的标记。另一方面,使用单独II型标记的结果与使用这两种类型标记的结果之间未观察到显著差异。图16B是图1650,其绘示了一种分析,所述分析对使用52个I型、52个II型以及两种类型(各52个)标记进行甲基化解卷积的准确性进行了比较。针对已经接受肝脏移植(LTP1到LTP5)的患者且针对已经接受骨髓移植(BMT1到BMT3)的患者,绘示通过甲基化解卷积所得到的移植器官对血浆DNA的贡献百分率与供者特异性SNP等位基因所产生的值之间的差异。针对每一患者,在左侧、在中间和在右侧用方框分别绘示使用单独I型标记、单独II型标记和两种类型标记的甲基化解卷积结果。使用单独I型标记的分析的偏差大于使用单独II型标记,或这两种类型的标记。另一方面,使用单独II型标记的结果与使用这两种类型标记的结果之间未观察到显著差异。图17A是图1700,其绘示了一种分析,所述分析对使用26个I型、26个II型以及两种类型(各26个)标记进行甲基化解卷积的准确性进行了比较。针对已经接受肝脏移植(LTP1到LTP5)的患者且针对已经接受骨髓移植(BMT1到BMT3)的患者,绘示通过甲基化解卷积所得到的移植器官对血浆DNA的贡献百分率与供者特异性SNP等位基因所产生的值之间的差异。针对每一患者,在左侧、在中间和在右侧用方框分别绘示使用单独I型标记、单独II型标记和两种类型标记的甲基化解卷积结果。使用单独I型标记的分析的偏差大于使用单独II型标记,或这两种类型的标记。另一方面,使用单独II型标记的结果与使用这两种类型标记的结果之间未观察到显著差异。图17B是图1750,其绘示了一种分析,其对使用13个I型、13个II型以及两种类型(各13个)标记进行甲基化解卷积的准确性进行了比较。针对已经接受肝脏移植(LTP1到LTP5)的患者且针对已经接受骨髓移植(BMT1到BMT3)的患者,绘示通过甲基化解卷积所得到的移植器官对血浆DNA的贡献百分率与供者特异性SNP等位基因所产生的值之间的差异。针对每一患者,在左侧、在中间和在右侧用方框分别绘示使用单独I型标记、单独II型标记和两种类型标记的甲基化解卷积结果。使用单独I型标记的分析的偏差显然大于使用单独II型标记或这两种类型的标记。另一方面,使用单独II型标记的结果与使用这两种类型标记的结果之间未观察到显著差异。总体上,II型标记提供的结果优于I型标记,这是令人吃惊的,尤其是此前研究中聚焦于I型标记。我们的结果还表明更多标记提供更大的准确度。E.不同准则的影响如上文所述,可以利用各种准则来鉴定不同类型的标记。举例来说,可以根据特定组织中的甲基化水平不同于所有组织的平均甲基化水平(例如至少相差特定的阈值,如3SD)来鉴定I型标记。并且,对于II型标记来说,使用某种变异和最大差异的准则。下述章节表明鉴定标记的不同准则的准确度。1.准则不太严格的标记的表现我们使用不同组织间具有不同变异率的标记对甲基化解卷积分析的表现进行比较。基于使用选择准则不同的两组标记,测定15位孕妇中的胎盘对血浆DNA的贡献。两组标记包括如此前章节中所述的I型标记。然而,II型标记的选择准则不同于两组标记。I组标记包括所有5820个II型标记,其满足甲基化密度CV>0.25的准则且各组组织的最大甲基化密度与最小甲基化密度之间的差异超过0.2。对于II组标记来说,CV要求是>0.15且各组组织的最大甲基化密度与最小甲基化密度之间的差异超过0.1。此组标记中存在8,511个II型标记。图18A是图1800,其根据本发明的实施例绘示了使用具有不同选择准则的标记所推知的胎盘对血浆DNA的贡献。竖轴对应于使用II组标记所推知的胎盘贡献。横轴对应于使用I组标记所推知的胎盘贡献。基于使用不同选择准则的两组标记的胎盘贡献结果之间存在良好相关性(r=0.99,皮尔逊相关系数)。因此,可以利用CV>0.15和各组组织的最大甲基化密度与最小甲基化密度之间的差异超过0.1的要求来获得良好准确度。2.同类型组织内的甲基化水平变异的影响为了调查同类型组织(例如来自不同个体)之间的标记甲基化水平的变异是否会影响解卷积分析的表现,我们分析来自两个怀孕个例的胎盘组织。鉴定两个类别的甲基化标记。具体地说,两个类别是基于其甲基化水平在两个胎盘组织中的相似性加以鉴定。i类标记具有10%或更低的甲基化密度。ii类标记在两个胎盘组织之间具有高度变异率(甲基化密度差异超过10%)。图18B是图1850,其绘示了在同类型组织中使用具有低变异率(i类)和高变异率(ii类)的标记进行血浆DNA解卷积的准确性。进行血浆DNA解卷积以测定15位孕妇的胎盘对血浆DNA的贡献。对于每个标记来说,使用两个胎盘组织的甲基化密度平均值表示分析中的胎盘的甲基化水平。在使用i类和ii类标记的每一个解卷积分析中,使用总共1024个标记。基于胎儿特异性SNP等位基因的比例,进一步测定血浆中的胎盘源DNA的量。然后根据基于胎儿特异性SNP等位基因的结果,对通过基于i类和ii类标记的甲基化解卷积分析所推导的贡献百分率进行比较。所得到的胎盘贡献相对于基于胎儿特异性等位基因所估算的值的中值偏离分别是2.7%和7.1%(使用i类和ii类标记)。因此,利用组织甲基化水平的个体间变异较低的i类标记在甲基化解卷积分析中得到更好的准确度。当使用同类型组织(ii类)内具有高度变异率的标记时,观察到甲基化解卷积结果与胎儿特异性等位基因分析结果之间存在显著较高的差异(P<0.0001,威尔科克森符号秩检验(Wilcoxonsign-ranktest))。换而言之,使用同类型组织内具有低变异率的标记会提高甲基化解卷积分析的准确度。因此,可基于同类型组织内的变异率来选择标记,例如(但不限于)CV值以及同类型组织的最大甲基化密度与最小甲基化密度之间的差异值。IV.根据贡献升高来鉴定组织中的疾病在利用所测定的贡献率的一种应用中,实施例可以相对于参考水平来检测特定组织类型的异常贡献率。在一个实施例中,参考水平可以对应于所述组织类型健康的生物体中所确立的值。在另一个实施例中,参考水平可以对应于利用不同尺寸范围的游离DNA分子所测定的贡献率。A.相对于健康百分率升高的百分率实施例可以检测到特定组织类型的所测定贡献率高于对健康生物体通常所预期的贡献率。特定组织类型的贡献率升高起因于组织发生病变且因此释放更多的游离DNA分子。举例来说,作为细胞凋亡或其它细胞机制的结果,病变的器官会释放更多的游离DNA分子。1.确定不明原发癌的组织起源在此前研究中,已经证明可以在癌症患者的无细胞血浆中检测到肿瘤相关的DNA变化。举例来说,可以在癌症患者的血浆DNA中检测到癌症相关的染色体拷贝数变化和癌症相关的全面低甲基化。因此,对血浆DNA的分析潜在地适用于筛选表面上健康个体中的癌症(Chan等人,《美国国家科学院院刊》2013;110:18761-8和Chan等人,《临床化学》2013;59:211-24)。检测血浆中的癌症相关特征之后,还重要的是确定原发肿瘤的位置。在此,我们提出肿瘤细胞会展现其所来源的原发组织的一些DNA甲基化特征。我们推测肿瘤源DNA的甲基化特征比其它组织更类似于原始起源组织。因此,在血浆中的肿瘤源DNA存在下,肿瘤所来源的组织对血浆DNA的贡献明显增加。因此,癌症患者血浆DNA中的组织特异性DNA甲基化模式的分析适用于指出原发肿瘤的部位。在这个实例中,我们分析了上文所论述的10位HCC患者、两位肺癌患者和一位结肠直肠癌患者的血浆DNA。分析中使用不同器官的甲基化模式。然而,由于在癌症筛选情形中,通常无法获得肿瘤组织用于甲基化分析,因此分析中不包括肿瘤组织的甲基化模式。图19是表1900,其根据本发明的实施例、基于器官特异性甲基化模式分析绘示了各种癌症患者和健康个体的不同组织对血浆DNA的贡献。与健康个体平均值相比,10位HCC患者血浆中有9位的肝脏贡献升高。肺癌患者和结肠直肠癌患者中的肺和结肠的贡献分别升高。因此,病变组织对应于异常贡献率。图20绘示了表2000,所述表根据本发明的实施例绘示了每位癌症患者的不同器官的贡献与四位对照个体的平均值的比较情况。贡献是用贡献率相对于四位对照个体的平均值的差异来表示。正值和负值分别表示特定器官的贡献增加和降低。在每位患者中,粗体数字表示相较于对照个体的最大增量。相较于四位对照个体,10位HCC患者中有8位HCC患者的肝脏贡献具有最大增幅。对于两位肺癌患者来说,肺的贡献展示最大增幅。对于结肠直肠癌患者来说,最大增幅来自结肠。这些结果表明血浆中的组织特异性甲基化模式分析可以适用于确定原发癌隐蔽的癌症起源。图21A是图2100,其根据本发明的实施例绘示了利用甲基化标记所估算的HCC和健康对照个体的肝脏对血浆DNA的贡献。相较于健康对照个体,肝脏对血浆的贡献在HCC个体中显著升高。因此,贡献率可以用作样品测量值,其中可以对测量值与阈值(例如约8%)进行比较以鉴定升高的疾病风险。与阈值的比较可以提供组织类型是否发生病变的分类,其中分类可以是发生病变的组织的不同机率水平。提供其它实例以便利用癌症检测所应用的甲基化解卷积来分析血浆DNA。为了证明此现象,分析29位肝细胞癌(HCC)患者、四位肺癌患者和一位结肠直肠癌患者的血浆DNA。招募三十二位健康个体作为对照,如图6的表600中所示。其中,26位HCC患者、4位肺癌患者和32位对照者的血浆DNA全基因组亚硫酸氢盐测序结果已经报道于此前研究中(Chan等人,《美国国家科学院院刊》2013;110:18761-8)。在这些实例中,血浆DNA的甲基化特征是利用亚硫酸氢盐测序来测定。也可以使用其它甲基化检测方法,例如(但不限于)最后一章中所提及的那些方法。图21B是图2150,其绘示了健康对照者与HCC患者之间,肝脏所贡献的血浆DNA的百分率,如根据本发明的实施例所推知。肝脏所贡献的血浆DNA百分率在HCC患者中显著高于对照个体(P<0.001,曼-惠特尼秩和检验(Mann-Whitneyrank-sumtest))。图2150提供了能够对组织贡献率与参考值进行比较以鉴定组织病变状态的其它证据。图22A和22B绘示了根据本发明的实施例、通过非怀孕健康对照者与肺癌或结肠直肠癌患者之间的比较所推知的(A)肺和(B)结肠的贡献百分率。图22A是图2200,其绘示了肺所贡献的血浆DNA的百分率在肺癌患者中显著高于(P=0.002,曼-惠特尼秩和检验)对照个体。图22B是图2250,其绘示了肺癌患者结肠所贡献的血浆DNA的百分率高于所有对照个体。这些数据表明,利用甲基化解卷积分析对血浆DNA进行分析适用于鉴定癌症起源组织(例如在患者已经鉴定为可能患有癌症之后)及用于筛选患者以首先鉴定组织疾病状态。图23是表2300,其根据本发明的实施例绘示了癌症患者间的血浆DNA组织映射分析。甲基化解卷积指出,肝脏对HCC和对照个体血浆的中值贡献百分率分别是12.9%(四分位区间:8.7%-32.9%)和5.5%(四分位区间:4.6%-7.1%)。2.基于升高的贡献来检测疾病状态的方法图24是一个流程图,其根据本发明的实施例图解说明了分析游离DNA分子的DNA混合物的方法2400,以基于组织对DNA混合物的贡献率提高来鉴定所述组织的疾病状态。生物样品包括来自多个组织类型(包括第一组织类型)的游离DNA分子的混合物。在步骤2410,鉴定N个基因组位点用于分析。N个基因组位点可以具有各种属性,例如如上文所述。举例来说,N个基因组位点可以仅包括I型或II型位点,或两者的组合。步骤2410可以按与图1的步骤110类似的方式进行。在步骤2420,接收包括来自M种组织类型的游离DNA分子混合物的生物样品。步骤2420可以按与图1的步骤130类似的方式进行。在步骤2430,分析生物样品中的游离DNA分子以鉴定其在与生物体对应的参考基因组中的位置。步骤2430可以按与图1的步骤140类似的方式进行。所分析的游离DNA分子可以是短DNA片段,其可以在DNA片段数目更少的情况下提供足够的准确度,如下述章节IV.B中所解释。在步骤2440,利用各自位于参考基因组的N个基因组位点中的任一个的游离DNA分子测量N个基因组位点的N个混合甲基化水平。可以针对N个基因组位点中的每一个测量一个混合甲基化水平。步骤2440可以按与图1方法100的步骤150类似的方式进行。因此,可以使用测量DNA分子甲基化水平的任何技术。在一些实施例中,DNA分子甲基化水平的测量可以使用甲基化感知测序结果,其也可以用于测定DNA分子的位置。在步骤2450,利用N个第一甲基化水平测定第一组织类型在混合物中的第一贡献率。在一些实施例中,步骤2450可以经由图1方法100的步骤160和170进行。从而可以同时测定一组M个组织类型的贡献率。步骤2450可以利用N个基因组位点的N个问题特异性甲基化水平,其针对M个组织类型中的每一个所测定,例如如图1方法100的步骤120。在步骤2460,计算第一贡献率与参考贡献率之间的分离值。举例来说,分离值可以包括第一贡献率与参考贡献率的差异或比率。分离值可以包括其它因数,且可以使用贡献率的函数差值。参考贡献率可以利用第一组织类型健康的生物体的样品测定。在步骤2470,可以对分离值与阈值进行比较以确定第一组织类型是否患有疾病状态的分类。如本文结果所示,混合物中的特定组织类型的量出现统计显著增加指示疾病状态。如果总贡献限制为1(即,100%),那么特定组织类型的增加将伴随混合物中的一或多种其它组织的相应降低。因此,可以对混合物中的第一组织类型的第一量(例如贡献率)与阈值量进行比较,以确定第一组织类型是否患有疾病状态的分类。在一个实施例中,基于第一组织类型健康的第一组生物体与第一组织类型发生病变的第二组生物体的混合物中的第一组织类型的量来确定阈值。病变生物体可以患有所测试的疾病,例如癌症。举例来说,第二组生物体的第一组织类型可以患有癌症。作为另一实例,第二组生物体可以移植已经排斥的第一组织类型。对于移植器官来说,疾病状态的鉴定可以对应于生物体是否排斥第一组织类型的分类,其中排斥反应是疾病状态。3.全身性红斑狼疮(SLE)为了进一步说明血浆DNA甲基化解卷积分析的潜在效用,我们分析九位SLE患者的血浆DNA。这些患者的SLE疾病活动性指数(SLEDAI)小于8,表明其疾病相对不活跃。对这些八位患者进行血浆DNA甲基化解卷积。图25是表2500,其根据本发明的实施例绘示了通过甲基化解卷积而得到的九位SLE患者的不同器官对血浆DNA的贡献百分率。与其它SLE患者相比,患者8及患者9的肝脏贡献升高。患者8患有药物诱发性肝炎,其具有235U/L的升高的丙胺酸转胺酶(ALT)活性。患者9患有累及肝脏的播散性肺结核。这些结果表明血浆DNA甲基化解卷积分析能够鉴定所影响器官的病理学。4.鉴定与所检测疾病有关的组织类型发现增加百分率大时,此前章节自动地确定组织类型为鉴定疾病的一部分。如果利用其它方式鉴定疾病,那么特定组织类型的较小增加可以允许鉴定组织类型,即使所述增加大不足以表示疾病状态本身。举例来说,如果如上鉴定出癌症,那么上述分析可以鉴定所累及的组织。针对所检测癌症鉴定组织类型的实施例的进一步描述提供于章节V中。B.利用甲基化解卷积进行的尺寸选择作为鉴定贡献率相对于得自健康组织的值升高的替代方案或除其之外,实施例可以分析对不同尺寸游离DNA分子的贡献率。当另外进行时,可以鉴定某些组织类型具有升高的贡献率,且尺寸分析可以证实组织类型是否发生病变。关于游离DNA分子的尺寸,已经证明胎源DNA的尺寸分布比孕妇血浆中的母源DNA短。另外,肿瘤源DNA的尺寸分布比癌症患者中的来源于非恶性组织的DNA的尺寸分布短(Jiang等人,《美国国家科学院院刊》2015;112:E1317-25)。就此而言,长DNA片段和短DNA片段的选择性分析能够鉴定短游离DNA分子从特定组织中的富集。相应地,升高的准确度可以通过分析特定尺寸的DNA片段来获得。举例来说,在罹患肝癌的患者中观察到肝脏对血浆DNA的贡献升高。已经证明来源于肝癌的血浆DNA分子比来源于非恶性组织的血浆DNA短(Jiang等人,《美国国家科学院院刊》2015;112:E1317-25)。因此,与分析长DNA分子时相比,分析短DNA分子时肝脏贡献较高的观察结果进一步支持肝脏贡献的升高与患者中肝癌的存在相容。1.结果利用成对端测序方案对三个母体血浆样品和两个来自癌症患者的血浆样品进行测序,以便可以确定每种血浆DNA分子两端的最外面核苷酸在参考人类基因组中的座标。然后根据两端的核苷酸的座标推导每种血浆DNA分子的尺寸。为了说明血浆DNA组成是否不同(当选择性地分析短或长DNA分子时),我们已经任意地使用150bp截止值来定义长DNA分子和短DNA分子。尺寸截止值的其它实例包括70bp、75bp、80bp、90bp、100bp、110bp、120bp、130bp、140bp、160bp、170bp、180bp、190bp和200bp。除长度之外,还可以使用质量作为尺寸的量度。作为用于质谱的一个实例,较长分子将具有更大的质量(尺寸值的一个实例)。长度是尺寸的另一实例,例如如利用碱基对所量度。尺寸选择也可以利用物理方法进行,如凝胶电泳或过滤或尺寸选择性沉淀或杂交。下述结果表明尺寸分析可以与经由甲基化解卷积进行的血浆DNA组织贡献分析组合使用。在一些实施例中,血浆DNA的甲基化解卷积可以聚焦于血浆DNA的特定尺寸范围。当来自非造血组织的DNA分子具有较短的尺寸分布时,短DNA片段的选择性分析可以对目标器官所释放的DNA进行更具成本效益的分析。举例来说,为了确定对接受肝脏移植的患者中的所移植肝脏是否有显著损伤,可以仅对短DNA片段进行甲基化解卷积。当选择性地分析短DNA片段时,由于非造血组织对血浆DNA的贡献率较高,因此可以在分析较少游离DNA分子的情况下获得相对于参考值的统计差值。举例来说,在游离DNA分子较少的情况下,由于来自非造血组织的游离DNA分子浓度较高,因此较高贡献率导致贡献率出现可检测到的变化(即,变化高于阈值)。相应地,方法2400中所分析的游离DNA分子可以低于尺寸截止值,从而可以在游离DNA分子较少的情况下提供期望的准确度。在这种情况下,肝脏贡献的增加可以表示所移植肝脏中的细胞死亡增加。图26A是图2600,其根据本发明的实施例绘示了所测定的三位孕妇(M6941p、M7171p和M396p)的胎盘对不同长度的游离DNA分子的贡献。与涉及所有血浆DNA的不使用尺寸选择的分析相比,仅当所分析的短血浆DNA片段<150bp时,胎盘对血浆DNA的贡献较高。相反,与涉及所有血浆DNA的不使用尺寸选择的分析相比,仅当所分析的长血浆DNA片段≥150bp时,胎盘对血浆DNA的贡献较低。这些结果符合比母源DNA短的胎盘源DNA(基因型与胎儿相同)的尺寸分布。此类结果表示实施例可以用于检测特定组织类型中的病状。图26B是表2650,其根据本发明的实施例绘示了所测定的移植患者的非造血组织对不同长度的游离DNA分子的贡献。将已经接受肝脏移植的五位患者(LT患者)的测序读数混合在一起用于分析。作为对照,将四位健康对照者的测序读数混合在一起用于此分析。我们观察到与涉及所有血浆DNA的不使用尺寸选择的分析相比,仅当所分析的短血浆DNA片段<150bp时,非造血组织的贡献比例升高。与涉及所有血浆DNA的不使用尺寸选择的分析相比,仅当所分析的长血浆DNA片段≥150bp时,贡献比例降低。此类结果还表示实施例可以鉴定器官中的病状。虽然实施例并非典型地用于鉴定移植器官,但是实施例可以监视不同尺寸的贡献率之间的分离值(例如差值或比率)。当分离值增大时,可以鉴定所移植的器官出现问题。图27A是图2700,其根据本发明的实施例绘示了所测定的移植患者的肝脏对不同长度的游离DNA分子的贡献。还针对健康对照个体和已经接受肝脏移植的患者分析肝脏的贡献比例。相对于涉及所有血浆DNA的不使用尺寸选择的分析,当分析短DNA片段时,肝脏的贡献比例升高,且当分析长DNA片段时,肝脏的贡献比例降低。与分析长DNA片段时相比,分析血浆中的短DNA片段时,肝脏的贡献更高。另外,差异的量大于非造血组织,包括除肝脏之外的其它组织。此类结果进一步说明能够鉴别患有与较短游离DNA分子的增加有关的病状的组织。图27B是图2750,其根据本发明的实施例绘示了所测定的HCC患者的肝脏对不同长度的游离DNA分子的贡献。分析两位HCC患者中的肝脏的贡献比例。相对于涉及所有血浆DNA的不使用尺寸选择的分析,当分析短DNA片段时,肝脏的贡献比例升高,且当分析长DNA片段时,肝脏的贡献比例降低。相应地,实施例可以分析长游离DNA分子与短游离DNA分子的贡献率之间的分离以鉴定发生病变的组织。可以针对一组组织类型中的每一个确定此类分离值。如果特定组织类型的特定分离值高于阈值,那么组织类型可以归类为对应于病变状态。正如可以看到的,正常生物体的差异仅是百分之几,其中在HCC病例中,差异接近8%或更多。2.方法图28是一个流程图,其根据本发明的实施例图解说明了一种分析游离DNA分子的DNA混合物的方法,以基于组织对不同尺寸的游离DNA分子的DNA混合物的差异贡献率来鉴定所述组织的疾病状态。生物样品包括来自多个组织类型(包括第一组织类型)的游离DNA分子的混合物。在步骤2810,分析来自生物样品的多个游离DNA分子。步骤3910可以按与图1方法100的步骤140类似的方式进行。举例来说,可以分析至少1,000个游离DNA分子以测定游离DNA分子所处的位置,且可以如下文所述测量甲基化水平。另外,可以测量多个游离DNA分子中的每一个的尺寸。可以用多种方式测量尺寸。举例来说,可以对游离DNA分子进行测序(例如使用甲基化感知测序)以获得序列读数,且尺寸可以对应于序列读数的长度。可以将序列读数与参考基因组进行比对以测定游离DNA分子所处的位置。在一个实施方案中,测序包括对每一个游离DNA分子的两个末端进行测序,且比对包括对两个末端进行比对。可以基于两个末端与参考基因组的比对来测定多个游离DNA分子的尺寸。位置和尺寸的测定可以按不同程序进行,例如可以进行物理分离,且然后可以测定位置(例如使用测序或杂交探针)。物理分离方法的实例包括凝胶电泳、过滤、尺寸选择性沉淀或杂交。可以先进行物理分离方法,随后分析游离DNA分子以测定其位置。在一个实施方案中,可以使用杂交探针来测定位置。在其它实施例(例如测序)中,可以测定多个游离DNA分子中的每一个的尺寸。在步骤2820,鉴定多个游离DNA分子,其各自位于对应于生物体的参考基因组的N个基因组位点中的任一个。只要游离DNA分子包括N个基因组位点之一,则可以包括其在内。N个基因组位点可以按不同方式且使用不同准则来鉴定,如本文所述。可以使用章节II中所述的技术。N是整数,其可以大于或等于10。在步骤2830,鉴定尺寸在第一尺寸范围内的第一组多个游离DNA分子。第一尺寸范围可以对应于任何尺寸范围,例如小于指定的长度、大于指定的长度,或两种尺寸之间。第一组可以如下鉴定:通过物理方法(例如如本文所述)或通过了解每个DNA分子的尺寸且用计算机鉴定其。在步骤2840,使用第一组多个游离DNA分子测量N个基因组位点的N个第一混合甲基化水平。可以针对N个基因组位点中的每一个测量一个第一混合甲基化水平。步骤28400可以按与图1方法100的步骤150类似的方式进行。在步骤2850,利用N个第一甲基化水平测定第一组织类型在混合物中的第一贡献率。在一些实施例中,步骤28500可以经由图1方法100的步骤160和170进行。从而可以同时测定一组M个组织类型的贡献率。在步骤2860,鉴定尺寸在第二尺寸范围内的第二组多个游离DNA分子。第二尺寸范围不同于第一尺寸范围。第二尺寸范围可以对应于任何尺寸范围,例如小于指定的长度、大于指定的长度,或不进行尺寸选择(即,所有尺寸),只要其相对于第一尺寸范围存在差异。当第二尺寸范围无尺寸选择时,第一尺寸范围将为第二尺寸范围的子集。在一些实施例中,两种尺寸范围不重叠,然而在其它实施例中可以存在重叠。尺寸范围的中心不处于相同的尺寸上,而是存在潜在不重叠的偏移。在一个实施例中,第一尺寸范围小于150个碱基且第二尺寸范围是150个碱基及更高。在步骤2870,使用第二组多个游离DNA分子测量N个基因组位点的N个第二混合甲基化水平。可以针对N个基因组位点中的每一个测量一个第二混合甲基化水平。步骤2870可以按与步骤2840类似的方式进行。在步骤2880,利用N个第二甲基化水平测定第一组织类型在混合物中的第二贡献率。步骤2880可以按与步骤2850类似的方式进行。在步骤2890,计算第一贡献率与第二贡献率之间的分离值。分离值的实例描述于本文中且包括差值或比率。如果一种组织类型对混合物贡献了相对更多的短DNA分子,那么尺寸范围越短,则贡献率越高。在步骤2895,可以对分离值与阈值进行比较以确定第一组织类型是否患有疾病状态的分类。分类可以指出当分离值超过阈值时,第一组织类型具有疾病状态。疾病状态可以鉴定组织因释放较短游离DNA分子的量不相称而出现的某种错误(例如癌症)。阈值可以定义为负值数字或可以确定绝对值。在一些实施例中,基于针对第一组织类型健康的第一组生物体与第一组织类型发生病变的第二组生物体的混合物所确定的分离值来确定阈值。不同分类可以解释分离值超过阈值的程度。相应地,可以使用多个阈值,如本文所述的任何方法中可以使用的。V.鉴定对应于拷贝数失常的组织拷贝数失常对应于染色体区域(例如整个染色体或染色体的一部分)中的扩增和缺失。拷贝数失常(CNA)存在于多种肿瘤中且因此可以指示癌症或其它疾病的存在。通过检测展现CNA的区域来鉴定癌症的其它细节可以见于美国专利第8,741,811号,所述专利并入本文供参考。然而,从CNA分析中可能无法严格地获知肿瘤起源。实施例可以利用甲基化解卷积鉴定对应于拷贝数失常的游离DNA分子的起源。实施例也可以利用甲基化解卷积测试特定的染色体区域。举例来说,血浆是由从体内多个组织释放的DNA组成。通过对血浆DNA使用全基因组亚硫酸氢盐测序,我们已经获得这些组织对循环DNA池的贡献。组织贡献者和其相对比例是通过利用代表各种组织类型的DNA甲基化标志绘制参考图的生物信息学解卷积方法鉴定,如上文所述。我们在孕妇、癌症患者和移植接受者中验证此方法。实施例允许鉴定血浆DNA中所观察到的基因组失常的起源组织。此方法在产前测试、肿瘤学、移植监测和其它领域中具有许多的研究和诊断应用。A.拷贝数失常(CNA)的组织映射检测血浆中的拷贝数失常已经在无创产前测试的背景下使用(ChiuRWK等人(2008)《美国国家科学院院刊》105:20458-20463;ChiuRWK等人(2011)《英国医学杂志(BMJ)》342:c7401;BayindirB等人(2015)《欧洲人类遗传学杂志(EurJHumGenet)》数字对象标识符(doi):10.1038/ejhg.2014.282;以及NortonME等人(2015)《新英格兰医学杂志(NEnglJMed)》372:1589-1597)和癌症检测(LearyRJ等人(2012)《科学·转化医学(SciTranslMed)》4(162):162ra154;Chan等人《美国国家科学院院刊》2013;110:18761-8;HeitzerE等人(2013)《国际癌症杂志(IntJCancer)》133(2):346-356)。如果可以鉴定出拷贝数失常的起源组织,那么是高度有利的。无创产前检测亚染色体拷贝数失常(YuSCY等人(2013)《科学公共图书馆综合卷》8(4):e60968)时,其适用于鉴定血浆失常是否来源于(i)单独胎盘、(ii)单独母亲,或(iii)胎盘与母亲。癌症筛选时,其在临床上提供了很多信息以便能够鉴定癌症起源组织,随后进行诊断或治疗程序。在不同类型的癌症中通常观察到拷贝数失常。癌症相关的拷贝数失常可以在癌症患者血浆中检测到(Chan等人《临床化学》2013;59:211-24)。在癌症筛选的背景下,CNA的起源组织可能不明显。因此,如果可以鉴定CNA的起源组织,那么其是适用的。血浆DNA甲基化解卷积可以用于鉴定血浆CNA的起源组织。图29是一个流程图,其根据本发明的实施例图解说明了一种用于确定拷贝数失常的起源组织的方法2900。方法2900可以使用患者血浆进行且至少部分地使用计算机系统进行。在步骤2910,进行血浆DNA分析以鉴定展现拷贝数失常的区域。失常可以对应于过度表达或表达不足。在一些实施例中,可以将基因组分成各分组(例如1Mb分组),且可以测定来自特定分组的游离DNA分子的量(例如通过使序列读数与参考基因组的那个部分对应)。可以将特定分组的量归一化(例如相对于一个分组的平均量),可以鉴定过度表达或表达不足。除基于CNA分析鉴定区域之外,可以简单地选择一个区域以便在各种实施例中测试。举例来说,可以怀疑一个区域具有例如CNA,因为某些区域在肿瘤中可能具有失常。或者,在胎儿应用(下述)中,某些染色体区域通常可能具有失常。在步骤2915,未鉴定出CNA区域。在一些实施例中,方法2900在此时可以停止。在步骤2920,可以进行甲基化解卷积,例如如图1中所述。可以针对每个CNA区域进行甲基化解卷积。相应地,可以进行染色体区域特异性血浆DNA甲基化解卷积。在步骤2932,作为甲基化解卷积的结果,获得拷贝数增加的区域的组织贡献。在步骤2934,作为甲基化解卷积的结果,获得不具有CNA的区域的组织贡献。在步骤2936,作为甲基化解卷积的结果,获得具有拷贝数损失的区域的组织贡献。在步骤2940,可以对不同染色体区域的组织贡献进行比较。举例来说,可以测定这些不同组织贡献的分离值。对于任何两个区域来说,可以测定特定组织的分离值。分离值将在具有拷贝数增加的区域与不具有CNA的区域之间,在具有拷贝数增加的区域与具有拷贝数损失的区域之间,以及在不具有CNA的区域与具有拷贝数损失的区域之间。在步骤2950,可以基于针对所述组织的分离值较大的程度来鉴定起源组织的身份。贡献大的组织会释放出具有所测试的失常的游离DNA分子。对于本申请来说,有利的是具有散布于整个基因组中的甲基化标记。就此而言,II型甲基化标记由于其数目相对大于I型标记而尤其适用。在某些实施例中,可以进一步调整标记的选择准则,以便进一步增加可以利用的标记数目。在又其它实施例中,可以将I型标记与II型标记组合以进一步增加可以利用的标记数目。B.鉴定异常区域CNA分析可以通过多种方式进行,例如如美国专利第8,741,811号中所述。举例来说,可以将人类基因组(或其它类型生物体的基因组)分成约3000个不重叠的1Mb分组。可以测定与每一个1Mb分组对应的读数的数目。校正GC偏差(ChenEZ等人(2011)《科学公共图书馆综合卷》6(7):e21791)之后,可以计算每个分组的序列读数密度。对于每个分组来说,可以对测试个例的测序读数密度与参考对照个体的值进行比较。拷贝数增加及损失可以定义为分别比对照组平均值高及低3个标准差。相应地,鉴定展现拷贝数失常的第一染色体区域可以是基于位于第一染色体区域中的游离DNA分子的第一量。为了确定血浆中拷贝数失常的组织起源,可以使用位于展现此类失常于血浆中的基因组区域内的甲基化标记进行血浆DNA组织映射。在关于癌症患者的下述实例中,仅在失常影响至少30Mb的邻接染色体区域的情况下进行血浆DNA拷贝数失常的映射,以便有足够数目个甲基化标记可以用于映射。C.检测CNA起源的实例甲基化解卷积可鉴定血浆拷贝数失常的起源组织。举例来说,当在血浆中观察到拷贝数增加时,位于所影响基因组区域内的标记的甲基化解卷积应该揭示了失常的起源组织的贡献升高(相较于对不具有拷贝数失常的基因组区域所执行的相同分析)。反之,当在血浆中观察到拷贝数损失时,位于所影响基因组区域内的标记的甲基化解卷积应该揭示了失常的起源组织的贡献降低。在以下章节中,我们说明了将此概念用于怀有第21对染色体三体症所影响的胎儿的孕妇、HCC患者和罹患淋巴瘤的孕妇。在这些实例中,不要求已知的所鉴定区域具有CNA;且在那种情况下,所述技术可以用于确定所测试区域是否存在序列不均衡。1.胎儿异常图30A根据本发明的实施例绘示了携有第21对染色体三体症的孕妇的染色体特异性血浆DNA甲基化解卷积的分析的图解说明。具有第21对染色体三体症的胎儿将数量增加的携带胎盘甲基化标志的染色体21序列释放到其怀孕母亲的血浆中。因此,当使用存在于染色体21上的标记对血浆亚硫酸氢盐测序数据进行甲基化解卷积时,预期胎盘贡献(表示为)相较于使用存在于其它染色体上的标记所估算的胎盘贡献(表示为)来说升高。在此图解说明中,假定母体血浆中的胎儿DNA分率是20%。由于胎儿中存在染色体21的额外拷贝,因此当基于染色体21上的标记进行甲基化解卷积分析时,胎盘源DNA的贡献与使用一或多种参考染色体上的标记相比将提高50%。相应地,实施例可以使用甲基化解卷积方法、使用来自染色体21的游离DNA分子测定贡献率,从而得到胎盘组织的贡献率为30%。还使用来自一或多种参考染色体的游离DNA分子进行甲基化解卷积,从而得到胎盘组织的贡献率为20%。然后可以测定不同组织的贡献率的差异以检测染色体21是否具有序列不均衡(例如这个实例中的三染色体性)。在此,我们将ΔM表示为染色体21与一或多种染色体(表示为RefChr)的不同器官对血浆DNA的贡献的差值。ΔM=MChr21-MRefChr其中MChr21是组织对血浆DNA的贡献(基于染色体21上的标记)且MRefChr是组织对血浆DNA的贡献(基于参考染色体上的标记)。因此,ΔM是各自对应于不同组织的贡献差值的矩阵。因此,实施例可以计算:涉及甲基化解卷积的每个其它组织类型的其它ΔM值将以类似方式计算。如果胎盘是母体血浆中染色体21拷贝数升高的起源,那么与其它组织类型的值相比,预期胎盘的ΔM值最高。为了进一步说明这种技术,我们分析了5位各怀有第21对染色体三体症胎儿的孕妇的血浆。孕龄范围在13周到14周之间。在每一例的血浆DNA中均观察到染色体21的表示增加。我们对测序数据进行甲基化解卷积且针对多个组织类型计算ΔM值。图30B是图表3050,其根据本发明的实施例绘示了各怀有第21对染色体三体症(T21)胎儿的孕妇的不同组织间的染色体21的分离值ΔM。在五种个例中的每一种中,胎盘的ΔM值最高,表明拷贝数失常来源于胎盘。并且,即使此前尚未鉴定出染色体21中的CNA,胎盘组织的高ΔM值也指示胎盘组织的染色体21中存在失常。图31是图表3050,其根据本发明的实施例绘示了各怀有第21对染色体三体症(T21)胎儿的孕妇的不同组织间的其它染色体的分离值ΔM。除染色体21外的所有常染色体上的甲基化标记随机分成两组,亦即A组和B组。随机分组是使用计算机产生的一系列随机数(范围为0到1)来实施。与随机数小于0.5有关的标记分到A组,否则将其分到B组。在此分析中,A组包括来源于染色体1、2、4、5、6、8、12、14、15、17、22的标记且B组包括来源于染色体3、7、9、10、11、13、16、18、19、20的标记。血浆DNA组织映射是使用每一组标记执行。所示ΔM值表示特定组织对血浆DNA的贡献差异(使用A组和B组的标记)。可以看出,单一组织不能始终展示升高的ΔM值。血浆DNA甲基化解卷积分析也可以适用于确定CNA是否来源于母亲或胎儿,例如在使用母体血浆DNA分析对微缺失或微复制进行的无创产前测试中。最近,已经表明胎儿的微缺失或微复制可以使用母体血浆DNA分析(Yu等人,《科学公共图书馆综合卷》2013;8:e60968)检测到。然而,当在母体血浆DNA中检测到微缺失或微复制时,失常可以来源于母亲、胎儿或其两者。甲基化解卷积分析可以用于解决此问题。考虑孕妇正常且携有微复制的情形。如果我们对母体血浆DNA中的复制区域和其它正常区域进行染色体特异性甲基化解卷积,那么ΔM值对胎盘来说大部分为正,这表明有额外剂量的胎盘DNA在复制区域释放到血浆中。另一方面,在母亲是微复制携带者且胎儿正常的情形中,胎盘DNA对母体血浆的贡献在复制区域相对减小,原因是在复制区域,母体组织对血浆DNA的贡献大于胎儿。如果母亲和胎儿均是微复制携带者,那么母亲和胎儿的贡献比例在受影响和不受影响的染色体区域并非不同。对于涉及微缺失的情形来说,反向关系将适用。ΔM在不同情形中的预期变化展示于下表中。表3.孕妇和她胎儿中的拷贝数变化不同的情形中的预期ΔM值在某些实施例中,胎儿或母亲或这两者可以在不同区域中携带超过一个拷贝数失常。举例来说,胎儿可以在不同区域中携带微复制和微缺失。2.肝细胞癌(HCC)一些实施例也可以用于确定由肿瘤引起的原始CNA。在呈现时肿瘤部位不明确的患者中,血浆DNA的CNA的甲基化解卷积分析将适用于鉴定癌症起源。图32A根据本发明的实施例图解说明了癌症患者的血浆DNA的CNA区域的分析。在癌症患者中,预期拷贝数增加(即扩增)的基因组区域富集于从相应癌症的起源组织释放的DNA中。因此将观察到癌症起源组织在血浆中的贡献比例增加(表示为)。相反,预期拷贝数减少(即缺失)的基因组区域在从相应癌症的组织释放的DNA中耗乏。然后观察到癌症起源组织在血浆中的贡献比例降低(表示为)。类似于上述第21对染色体三体症实例,可以使用以下方程式定义值ΔM,其中,在不是癌症起源组织的组织中,拷贝数失常(即扩增或缺失)对血浆的贡献比例不存在任何系统性影响。因此,在此类分析中,与其它组织类型的ΔM值相比,癌症起源组织的ΔM值最高。在其它实施例中,可以通过比较展示扩增的基因组区域与展示正常拷贝数的区域来计算ΔM。在又其它实施例中,可以通过比较展示缺失的基因组区域与展示正常拷贝数的区域来计算ΔM。举例来说,分析七位HCC患者、一位肺癌患者和一位结肠直肠癌患者的血浆DNA。所有这些九位患者的血浆中已检测到CNA。为了确定血浆中所检测到的这些CNA的起源组织,对展现拷贝数增加及拷贝数损失的染色体区域进行甲基化解卷积。在上文所研究的HCC、肺癌和结肠直肠癌样品中,在7位HCC、1位肺癌和1位结肠直肠癌患者的血浆中观察到影响至少30Mb区域(即人类基因组的约1%)的拷贝数失常。独立地测定每种组织类型对基于展示扩增和缺失的基因组区域的血浆的贡献比例。计算每种组织类型在两组基因组区域之间的贡献差异且表示为ΔM,其中ΔM是组织类型差异的矩阵。因此,ΔM=MAmp-MDel其中MAmp是基于位于展现拷贝数增加的基因组区域中的标记表示组织贡献的阵列;且是MDel基于位于展现拷贝数损失的基因组区域中的标记表示组织贡献的阵列。图32B是图表3250,其根据本发明的实施例绘示了癌症患者的不同组织间的展现拷贝数增加的区域与展现拷贝数损失的区域之间的分离值ΔM。在这个实例中,ΔM值跨越癌症患者的不同组织。ΔM表示特定组织对血浆DNA的贡献在展现拷贝数增加的区域与展现拷贝数损失的区域之间的差异。在每种情况下,最高ΔM是用黄色、蓝色或绿色展示。其它ΔM值是用灰色展示。具有最高ΔM的组织视为拷贝数失常的起源组织。七位HCC患者、肺癌患者和结肠直肠癌患者的肝脏、肺和结肠分别对血浆DNA的贡献在具有拷贝数增加的基因组区域与具有拷贝数损失的基因组区域之间的差异(ΔM)最高。因此,甲基化解卷积分析正确地指示血浆样品中的CNA的起源组织。图33是图表3300,其根据本发明的实施例绘示了癌症患者的不同组织间的随机选择基因组区域之间的分离值ΔM。作为对照,我们还使用血浆中未展现拷贝数失常的两组随机选择基因组区域进行相同分析。所示ΔM值表示特定组织对血浆DNA的贡献在两组不具有血浆DNA拷贝数失常的随机选择区域之间的差异。如在图33中可以看出,在此对照分析中,ΔM值与癌症起源组织之间不存在系统关系。3.患有淋巴瘤的孕妇除拷贝数失常之外,甲基化解卷积还可以应用于确定其它类型的基因组失常的起源组织,例如(但不限于)单核苷酸突变和易位。可以测定接近于基因组失常的区域的甲基化状况且与不受影响的区域的甲基化状况进行比较。预期基因组失常的起源组织在所影响的区域对血浆DNA展示较高贡献。图34A根据本发明的实施例绘示了患有并发淋巴瘤的孕妇的甲基化解卷积分析的图解说明。图38绘示了具有拷贝数增加的区域和不具有拷贝数增加的区域。为了证实血浆中所观察到的拷贝数失常的起源组织,可以独立地使用存在于在血浆中展示扩增的基因组区域(表示为)及展示正常拷贝数的区域(表示为)中的标记进行血浆甲基化解卷积:;图34A绘示了B细胞、胎盘和其它组织的贡献率图表。由于CNA的起源组织是滤泡性淋巴瘤,因此淋巴瘤所来源的组织类型(B细胞)将得到最高的ΔM值。为了进一步说明实施例的效用,我们分析了经诊断在怀孕早期期间患有复发滤泡性淋巴瘤的孕妇的血浆DNA。这位女性具有滤泡性淋巴瘤病史且接受过以治愈为目的的化学疗法。她随后怀孕,同时她的淋巴瘤处于临床缓解。在妊娠第11周期间,从孕妇收集血液样品,以便对胎儿染色体非整倍性进行无创产前测试。母体血浆DNA测序结果揭示总体异常。通过淋巴结的组织学检查和环钻活检来证实滤泡性淋巴瘤的复发。图34B是图3450,其绘示了从孕妇所收集到的试样间的拷贝数失常检测的全基因组DNA测序分析,所述孕妇经诊断在怀孕早期期间患有复发滤泡性淋巴瘤。图3450绘示了白细胞层、淋巴结活检体、治疗前血浆以及化学疗法开始后10周所收集的血浆样品的全基因组拷贝数分析。从内到外:治疗前血浆样品的白细胞层、淋巴结活检体、治疗之前收集的血浆样品和治疗之后收集的血浆样品。染色体G带图的最外层环是按顺时针方式绘示。每个点表示1Mb区域。绿点、红点和灰点分别表示具有拷贝数增加、拷贝数损失以及不具有拷贝数失常的区域。拷贝数从中心到外部是按升序排列。与其它染色体区域相比,圆点更接近于中心表示拷贝数损失。与其它染色体区域相比,进一步偏离中心的圆点表示拷贝数增加。在淋巴结活检体和治疗前血浆样品中检测到拷贝数失常,但是在治疗后血浆样品和治疗前血浆样品的白细胞层中则未检测到。淋巴瘤的拷贝数失常特征与治疗前血浆中的拷贝数失常特征之间存在高度相似性。治疗前血浆部分中存在拷贝数失常,但此类失常不存在于相同血液样品的血细胞部分中,表明血浆DNA异常来源于淋巴瘤相关的游离DNA,而非循环肿瘤细胞。对治疗前血浆样品进行全基因组亚硫酸氢盐测序,随后进行甲基化解卷积。在此患者中,血浆中展现拷贝数损失的邻接区域的尺寸无一者是30Mb或更高。因此,位于缺失区域内的甲基化标记数目对于组织映射分析来说是不够的。因此,未展现任何拷贝数失常的区域用作参考。图35A是表3500,其绘示了使用患有复发滤泡性淋巴瘤的孕妇的治疗前血浆样品,根据血浆DNA组织映射所测定的贡献率。淋巴细胞的血浆DNA的贡献比例是70.2%。B淋巴细胞的血浆DNA贡献是62.2%且T淋巴细胞贡献8%。图35B是图表3550,其绘示了患有并发滤泡性淋巴瘤的孕妇的不同组织的分离值ΔM。绘示此患者的治疗前血浆样品在不同组织间的ΔM值。B细胞展示最高ΔM值,表明拷贝数失常来源于B细胞。滤泡性淋巴瘤细胞来源于B细胞。可以看出,B淋巴细胞展示最高ΔM值,从而证实其是血浆中拷贝数失常的起源。4.癌症患者中的转移性病变这些基因组失常的甲基化解卷积可以特别适用于临床情形中,其中不确定肿瘤是所影响器官的原发癌,还是来源于另一个器官癌症的转移性病变。如上文所说明,器官受累于肿瘤将导致所影响器官对血浆的贡献改变。另外,通过甲基化解卷积对血浆DNA的CNA进行的分析适用于鉴定原发癌的组织起源。这两种类型的分析可以组合用于确定是否存在转移性病变。为了对此进行说明,下文论述三个假想实例:i.患有HCC(原发肝癌)的患者;ii.患有原发结肠直肠癌而无肝脏转移的患者;以及iii.患有原发结肠直肠癌伴肝脏转移的患者。肝脏的贡献血浆中的CNA的解卷积HCC患者增加来自肝脏的CNA无肝脏转移的结肠直肠癌患者正常来自结肠的CNA伴有肝脏转移的结肠直肠癌患者增加来自结肠的CNA表4.针对三位假想患者的血浆DNA甲基化解卷积分析的预期结果。对于HCC患者来说,肝脏中存在肿瘤将导致肝脏对血浆DNA的贡献增加。另外,由于癌症来源于肝细胞,因此与癌症有关的CNA的组织起源是肝脏。对于无肝脏转移的结肠直肠患者来说,由于未累及肝脏,因此预期肝脏对血浆DNA的贡献正常;且甲基化解卷积指示肿瘤来源于结肠。对于伴有肝脏转移的结肠直肠癌患者来说,肿瘤细胞侵入肝脏将导致释放到血浆中的肝脏DNA增加。由于癌症来源于结肠,因此CNA分析将指示失常来源于结肠。举例来说,对呈现有肝肿块的患者进行超声波扫描术研究。经过后续的临床调查,发现患者的结肠直肠癌转移到肝脏。对血浆进行甲基化解卷积。表5绘示了此患者展示结肠对血浆DNA的贡献增加。表5.在超声波扫描术研究中,具有肝肿块的患者的贡献率。图36A是图3600,其根据本发明的实施例绘示了对结肠直肠癌转移到肝脏的患者的血浆DNA进行的拷贝数失常分析。每个点表示1Mb区域。结果是按一组32位健康对照个体的血浆DNA的标准差相对于平均基因组表示的数目表示。位于两条黑线之间的灰点指示血浆DNA表示不偏离健康个体平均值。位于两条黑线之间的区域内部和外部的黑点指示那些区域在患者的血浆DNA中分别是表示不足或过度表示。然后使用解卷积分析对血浆DNA中过度表示和表示不足的区域进行分析以确定失常的组织起源。图36B是图表36B,其根据本发明的实施例绘示了患有结肠直肠癌和肝脏转移的患者的血浆DNA的拷贝数失常的甲基化解卷积分析。分析指出结肠的扩增区域与缺失区域之间的差异(ΔM)最大,表明失常最可能来源于结肠。因此,实施例能够鉴定引起肝肿块的原发癌。5.体细胞嵌合体细胞嵌合描述具有不同基因组构成的细胞在身体的某些组织中的存在。这来源于染色体隔离或DNA复制期间发生的错误,从而引起多种基因组失常,如染色体非整倍性、拷贝数变异(CNV)、基因组重排、单核苷酸变异,或重复扩增和微卫星不稳定性(Lupski,《科学》2013;341:358-9)。血浆DNA甲基化解卷积的实施例可以适用于鉴定体细胞嵌合所影响的组织。首先分析血浆DNA以表征基因组失常,例如CNA。然后,可以使用受影响区域和不受影响的另一个区域内的甲基化标记进行甲基化解卷积。通过比较这两组区域的血浆DNA的组成,可以测定ΔM。然后可以根据具有显著分离值(例如ΔM值)的组织来鉴定被体细胞嵌合影响的组织。6.各种病理学病状的检测和监测血浆DNA甲基化卷积可以用于检测和监测各种病理学病状,例如(但不限于)中风、心肌梗塞、自体免疫疾病和感染。举例来说,承认患者意识丧失且怀疑中风的临床诊断。脑的贡献升高可以适用于指示脑存在重大损伤。脑对血浆DNA的贡献升高可以通过比较患者的结果与健康对照个体的结果来断定。还可以利用贡献的升高程度来指示患者的预后。类似地,对于因临床症状而怀疑患有心肌梗塞或其它心脏疾病的患者来说,心脏的贡献可以用于指示诊断或预测患者的预后。可以利用一组健康对照个体的心脏对血浆DNA的贡献的值来确定截止值。在一个实施例中,截止值可以是健康对照个体的脑贡献的某个百分位,例如第90个、第95个或第99个百分位。在另一个实施例中,截止值可以设定为比对照个体平均值高2SD、2.5SD、3SD或3.5SD。血浆DNA甲基化解卷积还可以应用于鉴定呈现起源不明的败血症的患者的感染来源。由于细胞损伤增加,因此预期所感染的组织将更多DNA释放到血浆中。7.总结如上文所详述,实施例就检测以下对血浆的贡献来说已经验证:(i)胎盘(使用孕妇);(ii)肝脏(使用HCC患者和肝脏移植后的个体);(iii)白血细胞(使用骨髓移植接受者和在怀孕期间所诊断的淋巴瘤病例);(iv)得自肺癌病例的肺;以及(v)得自结肠直肠癌病例的结肠。因为血浆DNA通常被视为细胞死亡的标记,因此我们的方法可以作为通用方法用于评估不同组织类型中的细胞死亡现象。因此,除应用于产前测试、癌症检测/监测和移植监测之外,实施例还可以应用于医学的许多分支中用于研究细胞死亡或各种身体组织的损伤,例如中风、心肌梗塞、创伤、自体免疫病症、传染病等。另外,数据表明根据患者的生理学状态或潜在病理学将观察到血浆DNA池的组织组成的特征性扰动。能够鉴定在血浆中可以观察到的拷贝数失常的组织起源具有许多潜在的临床应用。举例来说,利用血浆DNA测序筛选癌症时,实施例可以鉴定癌症的可能起源组织,以便计划好进一步的诊断调查或治疗程序。作为另一实例,实施例非常适用于无创产前测试。使用第21对染色体三体症的检测作为模型系统,我们已经证明可以鉴定出胎盘为母体血浆中过量的染色体21的组织起源。癌症检测和无创产前测试的应用集中于罹患滤泡性淋巴瘤的孕妇病例中。我们观察到此孕妇的血浆中出现拷贝数失常(图34A)。血浆甲基化解卷积揭示了淋巴细胞对血浆的贡献非常高。B淋巴细胞是涉及滤泡性淋巴瘤病理学的细胞类型。因此,令人感兴趣的是实施例鉴定出B细胞(62.2%,图35A)而非T细胞为患者中的血浆DNA的主要贡献者。对使用来源于展示拷贝数失常增加相对于展示正常拷贝数目的基因组区域的甲基化标记所得的甲基化解卷积结果进行比较的ΔM分析进一步证实B细胞为拷贝数失常的来源(图35B)。这些结果因此完全符合滤泡性淋巴瘤的诊断。随着无创产前测试的临床效用增加和母亲年龄进一步提前的倾向,在此类测试的过程中可能将检测到越来越多的恶性疾病病例(OsborneCM等人(2013)《产前诊断(PrenatDiagn)》33(6):609-611;VandenbergheP等人(2015)《血液科柳叶刀(LancetHaematol)》2:e55-e65)。本文所述的实施例因此非常适用于进一步调查此类病例。在一些实施例中,可以进一步改进用于解卷积方法的甲基化标记的选择。在一种变化形式中,可以调整标记组以更多地关注对血浆DNA池的贡献不太显著的组织类型。这可以揭示出可以利用实施例监视的新病理生理学状况。除使用DNA甲基化标记之外,实施例还可以通过研究mRNA(NgEKO等人(2003)《美国国家科学院院刊(ProcNatlAcadSciUSA)》100:4748-4753;TsuiNBY等人(2014)《临床化学》60(7):954-962;KohW等人(2014)《美国国家科学院院刊》111(20):7361-7366)和微RNA(ChimSSC等人(2008)《临床化学》54(3):482-490;WangK等人(2009)《美国国家科学院院刊》106(11):4402-4407)来调查组织对循环核酸池的贡献。DNA甲基化和转录组学方法可以彼此协同发挥作用且得到不同类型的信息。在上述实例中,根据制造商说明书(Illumina)制备DNA文库且在HiSeq或NextSeq系统(Illumina)上测序。对于HiSeq来说,使用TruSeqSBS试剂盒v3(Illumina)进行76(单端模式)或76×2(成对端模式)个测序循环。对于NextSeq来说,使用NextSeq500高输出型v2试剂盒(Illumina)进行76×2个成对端测序循环。碱基识别之后,除去衔接序列和低品质碱基(即,品质分数<5)。然后通过甲基化数据分析流水线Methy-Pipe对呈FASTQ格式的微调读数进行处理。所有样品的基本测序参数(包括测序深度)概述于图37和图38的表3700中。D.用于测定序列不均衡的方法图39是一个流程图,其根据本发明的实施例图解说明了一种使用甲基化解卷积来分析生物体的生物样品的方法3900,以确定染色体区域是否展现序列不均衡。生物样品包括来自多个组织类型(包括第一组织类型)的游离DNA分子的混合物。方法3900至少部分地使用计算机系统进行。在步骤3910,分析来自生物样品的多个游离DNA分子。步骤3910可以按与图1方法100的步骤140类似的方式进行。举例来说,可以分析至少1,000个游离DNA分子以测定游离DNA分子所处的位置,且可以如下文所述测量甲基化水平。在步骤3920,鉴定第一组多个游离DNA分子。第一组中的每个DNA分子位于对应于生物体的参考基因组的第一染色体区域的N个基因组位点中的任一个。举例来说,一个DNA分子可以位于N个基因组位点中的第一个(例如具有与其比对的序列读数),且另一个DNA分子可以位于N个基因组位点中的第二个。两个DNA分子均包括于第一组中。N个基因组位点可以按不同方式且使用不同准则鉴定。可以使用章节II中所述的技术。N个基因组位点可以满足某些准则,如跨越组织和跨越个体的甲基化水平。可以基于其它样品的数据(例如数据库的甲基化分析)鉴定基因组位点。N是整数,其可以大于或等于10。N个基因组位点位于第一染色体区域中,第一染色体区域可以是邻接的或由非邻接子区域组成。第一染色体区域可基于CNA分析(例如如上文所述)而选择。举例来说,可以鉴定一个区域相对于其它区域具有DNA分子的过度表示或表示不足,其中所述分析可以潜在地使用与甲基化分析所用相同的生物样品。过度表示或表示不足表明拷贝数失常,且下述甲基化分析可以确定哪个组织是CNA起源。在步骤3930,使用第一组多个游离DNA分子测量N个基因组位点的N个第一混合甲基化水平。可以针对N个基因组位点中的每一个测量一个第一混合甲基化水平。步骤3930可以按与图1方法100的步骤150类似的方式进行。因此,可以使用测量DNA分子甲基化水平的任何技术。在一些实施例中,DNA分子甲基化水平的测量可以使用甲基化感知测序结果,其也可以用于测定DNA分子的位置。在步骤3940,利用N个第一甲基化水平测定第一组织类型在混合物中的第一贡献率。在一些实施例中,步骤3940可以经由图1方法100的步骤160和170进行。从而可以同时测定一组M个组织类型的贡献率。步骤3940可以利用N个基因组位点的N个问题特异性甲基化水平,其针对M个组织类型中的每一个所测定,例如如图1方法100的步骤120。在一些实施例中,N个组织特异性甲基化水平可能仅针对第一组织类型以及共同针对所有其它组织类型。因此,M可以有效地仅是2。如果第一组织类型是所关注的唯一组织类型,那么此通则不损失任何信息。其它组织类型的共同值可以利用每一个其它组织类型的独立值产生。在步骤3950,鉴定第二组多个游离DNA分子。第二组中的每个DNA分子位于对应于生物体的参考基因组的第二染色体区域的N个基因组位点中的任一个。第二染色体区域不同于第一染色体区域(例如不同染色体),且因此,K个基因组位点不同于N个基因组位点。K是整数,其可以大于或等于10。K和N的值也可以是不同的,且因此,K可以不等于N。步骤3950可以按与步骤3920类似的方式进行。第二染色体区域可以鉴定为不展现任何失常的区域。鉴定可以是基于测量来自生物体的样品,例如当鉴定第一染色体区域时,按类似方式,而非展示任何过度表示或表示不足。在其它实施例中,第二染色体区域可以鉴定为具有来自第一染色体区域的相反失常,其中所述失常可以假设来自相同组织类型。在又其它实施例中,可以基于典型的失常位置或其缺乏来鉴定第二染色体区域。举胎儿为例,非整倍性相对更常见地发生于染色体13、18和21,但对于其它染色体来说相对不常见。因此,其它染色体中的一或多种可以用作第二染色体区域。第二染色体区域可以是邻接的或相邻的或由非邻接的子区域组成。在步骤3960,使用第二组多个游离DNA分子测量K个基因组位点的K个第二混合甲基化水平。可以针对K个基因组位点中的每一个测量一个第二混合甲基化水平。步骤3960可以按与步骤3930类似的方式进行。在步骤3970,利用K个第二甲基化水平测定第一组织类型在混合物中的第二贡献率。步骤3970可以按与步骤3940类似的方式进行。在步骤3980,计算第一贡献率与第二贡献率之间的分离值。举例来说,分离值可以包括第一贡献率与第二贡献率的差异或比率。分离值可以包括其它因数,例如相乘因数。作为其它实例,可以使用贡献率的函数差,例如贡献率的自然对数(ln)的差值。在步骤3990,对第一分离值与阈值进行比较以确定第一组织类型的第一染色体区域是否具有序列不均衡的分类。分类可以是当分离值超过阈值时,第一组织类型的第一染色体区域具有序列不均衡。如此前章节中所述,较大分离值指出第一组织类型存在序列不均衡(例如拷贝数失常)。举例来说,如果第一贡献率比第二贡献率大阈值,那么可以确定第一染色体区域在第一组织类型中展现扩增。如果第一贡献率比第二贡献率小阈值,那么可以确定第一染色体区域在第一组织类型中展现缺失。在一个实例中,生物体怀有胎儿,且第一组织类型是胎盘组织,如章节V.C.1。因此,方法可以检测胎儿的第一染色体区域是否具有非整倍性。在另一个实例中,第一组织类型可以不是胎盘组织,即使生物体怀孕。此测试可以确定其它组织是否具有序列不均衡,例如如章节V.C.3。如上文所提及,第一染色体区域可以基于位于第一染色体区域的游离DNA分子的量鉴定为展现拷贝数失常。所述量相对于另一个区域(例如至少一个阈值)的过度表示或表示不足可以指示拷贝数失常。举例来说,位于第一染色体区域中的游离DNA分子的量可以是游离DNA分子的原始计数、游离DNA分子的累计长度,以及可以作为区域中每单位长度的计数测定的密度。区域一经鉴定用于测试,则可以确定M个组织类型的分离值。因此,对于第一和第二染色体区域中的每一个来说,M个组织类型中的每一个可以确定M个贡献率。可以对每一个分离值与阈值进行比较,以确定组织类型是否是起源。分离值可以表示超过一个组织类型展现序列不均衡,如V.C.4中。在一个实施例中,最大分离值可以鉴定是原发癌。如果生物体经鉴定在某些组织(例如非胎盘组织)的第一染色体区域中具有序列不均衡,那么生物体可以归类为某种组织具有某种癌症等级。癌症等级可以基于分离值的范围确定。癌症等级可以基于第一染色体区域的过度表示或表示不足的水平以及展现失常的染色体区域数目来进一步确定。在一些实施例中,可以测试第一组织类型中的多个区域的序列不均衡。如果第一组织类型的许多区域(例如超过截止值)展现序列不均衡,那么第一组织类型鉴定为起源可以具有更大的统计精确性。并且,如果测试许多区域,那么可以减小用于确定序列不均衡的阈值,其中使用具有序列不均衡的区域的数目截止值来提高特异性。因此,第一组织类型的第一染色体区域是否具有序列不均衡的分类可以基于具有超过阈值的相应分离值的不同染色体区域的数目。以此方式可以通过鉴定具有较小分离值的区域来提高灵敏度(否则的话可能检测不到)。阈值可以取决于截止值;截止值越高,阈值就越低,且反之亦然。生物体一经诊断具有某种癌症等级,则可以基于诊断来治疗生物体。也可以根据对疾病状态进行分类的其它方法进行治疗。举例来说,治疗可以包括手术、放射疗法或化学疗法。VI.靶向分析基于甲基化分析对组织贡献进行解卷积可以涉及测定CpG位点的甲基化状况。除使用非靶向亚硫酸氢盐测序测定DNA混合物(例如血浆DNA)的全基因组甲基化特征之外,还可以使用靶向方法研究所关注的CpG位点的甲基化状况或甲基化密度,或其它甲基化水平。可以靶向所关注的CpG位点,例如(但不限于)DNA杂交、微阵列、PCR扩增和甲基化特异性PCR。这些技术还可以组合使用。靶向方法可以增加关于个别CpG位点的甲基化信息而不会大幅度增加总体测序的量。靶向方法还可以提高检测组织对体液中的DNA贡献的灵敏度和/或特异性和/或精确度,尤其是与一或多种其它组织相比时贡献较少的组织。在一个实例中,所关注的区域可以通过杂交来富集,例如(但不限于)使用NimblegenSeqCap系统或AgilentSureSelect目标富集系统。在另一个实例中,可以设计尤其可捕捉亚硫酸氢盐转化DNA序列的杂交探针。然后可以对所关注区域富集的测序文库进行测序。使用此策略可以显著增加所关注区域的测序深度,其中所测序的样品中DNA分子数目与非靶向测序方法相同。作为另一实例,所关注的区域可以使用PCR扩增靶向。可以设计PCR引子以利用为甲基化解卷积分析提供信息的CpG位点扩增区域。可以分析扩增区域,例如(但不限于)针对总体甲基化水平使用大规模平行测序,包括单分子测序(如纳米孔测序或PacificBiosciences单分子实时系统)、实时PCR、数字PCR或质谱。在一个实施方案中,可以设计靶向甲基化序列或未甲基化序列的PCR引子。在此实施方案中,可以对甲基化和未甲基化DNA分子的量进行比较,以便测定信息性CpG位点(I型或II型甲基化标记)的甲基化水平。在另一个实施方案中,PCR引子仅与不具有差异性甲基化的区域(例如不具有CpG位点的区域)杂交。在这种情况下,可以扩增甲基化序列与未甲基化序列。然而,经扩增的扩增子含有CpG位点且然后可以测定每个经扩增的分子的甲基化状况,例如(但不限于)使用对甲基化或未甲基化序列具有特异性的荧光探针。或者,可以使用大规模平行测序或质谱来分析PCR产物。还可以应用各种实施例分析不同CpG位点的甲基化特征以便最大化分析的成本有效性。A.靶向I型与II型标记靶向I型与II型标记适用于提高甲基化解卷积分析的总体成本有效性,因为大量的所分析游离DNA分子对应于所使用的基因组位点。换而言之,为了得到相同数目个信息性DNA分子用于甲基化解卷积分析,与使用全基因组分析相比可以大幅度减小使用靶向方法测序的量。B.靶向I型标记和II型标记的全基因组分析当需要测定特定类型的组织的贡献且其它组织的贡献受到一般关注时,靶向I型标记和对II型标记的全基因组分析特别适用。虽然靶向I型标记与II型标记也可以实现此目标,但设计靶向两种类型标记的分析可能需要大量的努力。在此情形下,所关注的组织中发生差异甲基化的I型标记可以按靶向方式分析,以便可以更精确地测定其在DNA混合物(例如血浆DNA和尿DNA)中的甲基化水平。在一些实例中,I型标记所靶向的组织对血浆DNA池的贡献较少。使用I型标记靶向此类组织使得可以检测以及测量其对血浆DNA池的贡献的灵敏度增加。另一优势是可以调节使此类测量优化的浓度范围。作为说明,如果希望靶向向血浆中通常贡献极低含量DNA的组织A,可以使用多个靶向组织A的I型标记,例如使用10或100个标记。如果所述10或100个标记中仅一部分对于特定血浆样品来说呈阳性,那么可以进一步调整组织A对血浆的所测量贡献。当组织A对血浆的贡献非常低时,检测到血浆中对组织A具有特异性的标记的概率较低且通过一或多种统计函数(例如泊松分布(Poissondistribution))来控制检测率。在这种情况下,可以根据血浆中可检测到的I型标记的百分率推导出组织A对血浆DNA的相对贡献。其它组织的贡献可以使用II型标记测定。C.靶向II型标记以及I型标记的全基因组分析靶向II型标记和I型标记的全基因组分析可以适用于排除特定组织类型的贡献。举例来说,预期胎盘的贡献在分娩之后降到检测不到的水平。II型标记的靶向分析和对胎盘具有特异性的I型标记的全基因组分析可以适用于准确地测定不同组织器官的贡献以及排除胎盘对血浆DNA的贡献。这可以适用于排除此前怀孕的女性中持留的妊娠期产物。VII.不同的无细胞体液的甲基化解卷积A.尿液DNA还可以对尿液DNA进行DNA甲基化解卷积。此前研究已经证明游离DNA可以在健康个体和患有多种疾病的患者中检测到(Hung等人,《临床化学》2009;55:715-22;Chan等人,《临床癌症研究(ClinCancerRes.)》2008;14:4809-13;GarcíaMoreira等人,《临床生物化学(ClinBiochem.)》2009;42:729-31;Hoque等人,《国立癌症研究所杂志(JNatlCancerInst.)》2006;98:996-1004)。尿液中的游离DNA可以局部来源于肾和泌尿系统中的细胞(Hoque等人,《国立癌症研究所杂志》2006;98:996-1004)或经肾来源于血浆(Hung等人,《临床化学》2009;55:715-22;Chan等人,《临床癌症研究》2008;14:4809-13)。甲基化解卷积分析可以适用于鉴定局部和全身疾病。在一个实施例中,尿液DNA的甲基化解卷积可以用于监测已经接受肾移植的患者。此前已经表明在肾移植接受者中,在移植物排斥反应存在下,增加的DNA从移植的肾脏释放到尿液中(Zhong等人,《纽约科学院年鉴(AnnNYAcadSci.)》2001;945:250-7)。因此,肾脏对尿液DNA的贡献百分率的升高适用于指示肾排斥反应的存在。在另一个实施例中,可以使用尿液DNA解卷积来检测或监测尿道中恶性疾病的存在。可以根据对尿液DNA的贡献的升高来指示癌症的组织起源。举例来说,预期患有膀胱和前列腺癌的患者的膀胱和前列腺的贡献分别升高。还可以结合基因组失常(例如拷贝数失常和单核苷酸变异体)来进行甲基化解卷积,以发现基因组失常的组织起源。其它临床情形(如感染和创伤)还可以通过尿液DNA的解卷积来检测。在感染的情况下,利用甲基化解卷积,会看到从白细胞群相对于尿液DNA的浓度增大。也可以应用尿液DNA甲基化解卷积检测以及监测肾脏病症。举例来说,可以应用所述技术检测以及监测具有自体免疫起源的肾病。在一个实施例中,会看到所选白细胞群(例如淋巴细胞)对尿液DNA池的异常贡献。自体免疫相关肾脏病症的实例包括IgA肾病变和肾小球肾炎(由于全身性红斑狼疮)。作为另一实例,可以应用所述技术检测以及监测肾小球过滤屏障存在损伤的肾病。在此类情况下,预期尿液DNA的经肾组分会增加。在又另一个实施例中,可以利用尿液DNA甲基化解卷积检测肾脏恶性疾病,例如肾细胞癌症和肾盂移行细胞癌。在此情形下,还可以结合基因组失常(例如拷贝数失常和单核苷酸变异体)来进行甲基化解卷积,以发现基因组失常的组织起源。从两位处于怀孕第三个三月期的孕妇收集尿液样品。对于每个尿液样品来说,使用WizardPlusMiniprepsDNA提纯系统(Promega)从17mL尿液中提取DNA,如此前所述(Tsui等人,《科学公共图书馆综合卷》2012;7:e48319)。利用KAPADNA文库制备试剂盒(KapaBiosystems)制备DNA测序文库。然后使用EpiTect亚硫酸氢盐试剂盒(Qiagen)对尿液DNA测序文库进行2轮亚硫酸氢盐修饰。通过10个PCR循环来富集经衔接子连接的DNA分子。经亚硫酸氢盐处理的DNA文库在HiSeq2000仪器(Illumina)上以成对端形式进行75bp测序。将测序读数与人类参考基因组(hg19)比对。基于1013个I型和5820个II型标记的甲基化水平进行解卷积分析以测定不同器官对尿液DNA的贡献。病例1病例2肝脏6.67.3肺14.216.9结肠8.56.0小肠3.31.3胰脏15.812.6膀胱12.28.5肾上腺1.60.0食道17.88.1脂肪组织0.01.8心脏0.00.0脑8.46.5T细胞0.00.0B细胞0.06.6嗜中性白细胞7.419.3胎盘4.35.0表6.利用尿液样品测定的贡献率。表6绘示了两位孕妇的不同器官相对于尿液的贡献百分率。尿液DNA的4.3%和5%经推导来自胎盘。这符合此前的发现:胎儿DNA可以经肾传递到孕妇尿液中(Tsui等人,《科学公共图书馆综合卷》2012;7:e48319)。另外,在两个尿液样品中,膀胱还贡献了总DNA的12.2%和8.1%。每个尿液DNA分子的尺寸可以根据最外层核苷酸的基因组座标来推导。图42A是图4200,其根据本发明的实施例绘示了两位孕妇的尿液DNA的尺寸分布。作为比较,还绘示了五位孕妇的血浆DNA的尺寸分布。尿液DNA的尺寸分布显著短于血浆DNA的尺寸分布。这些发现指出,对短尿液DNA进行甲基化解卷积是可行的。图42B根据本发明的实施例绘示了尿液DNA中的不同染色体的基因组表示(GR)图4250。作为比较,还绘示了来自两位孕妇的血浆DNA样品的染色体的基因组表示。不同染色体的比例表示在尿液与血浆样品之间是类似的。与染色体Y比对的尿液DNA序列的0.063%和0.059%。这与两位孕妇均患有男性胎儿的事实相容。B.脑脊髓液(CSF)作为另一实例,还可以对从CSF中提取的DNA进行甲基化解卷积。增强的组织损坏可能与不同的颅内病理学有关,例如脑血管疾病、感染、癌症、自体免疫病症(例如多发性硬化症)和退行性病症(例如阿尔茨海默病(Alzheimer'sdisease)、帕金森病(Parkinson'sdisease)等。特定细胞类型对CSF中的DNA的贡献升高与那种特定细胞类型的细胞周转率增加有关且可以用于检测和监测(包括对治疗的响应)各种疾病。C.胸膜液和腹水液在另一个实例中,还可以对从胸膜液中提取的DNA进行甲基化解卷积。在罹患各种肺病变的患者中通常观察到胸膜积液。还在患有心脏衰竭、肾病的患者和患有肝病的患者中观察到胸膜积液。在此前研究中,已经表明有胸膜积液的患者的胸膜液中的DNA浓度的量度适用于将胸膜积液分类成通透型和渗出型(Chan等人,《临床化学》2003;49:740-5)。此分类适用于指示患者正罹患的可能病变。胸膜液DNA的解卷积适用于指示病变的组织起源。举例来说,在罹患恶性胸膜积液的患者中,胸膜液的解卷积可以指示胸膜积液是否归因于原发肺癌或另一器官的癌症转移到肺。另外,可以对展现不同类型的遗传失常(包括拷贝数失常和点突变)的区域进行甲基化解卷积,以便可以确定失常的组织起源。在又另一实例中,可以对从腹水液中提取的DNA进行甲基化解卷积。可以在各种病变(例如肝硬化、感染和恶性疾病)中观察到腹水。其还可以在患有心脏衰竭和肾病的个体中观察到。腹水液DNA的解卷积适用于指示病变的组织起源。具体地说,鉴定引起腹水的恶性疾病的起源。类似于胸膜液的分析,可以对展现不同类型的遗传失常(包括拷贝数失常和点突变)的区域进行甲基化解卷积,以便可以确定失常的组织起源。VIII.计算机系统在此提及的任何计算机系统都可以利用任何适合数目个子系统。此类子系统的实例在图42中是以计算机设备10绘示。在一些实施例中,计算机系统包括单个计算机设备,其中子系统可以是计算机设备的组件。在其它实施例中,计算机系统可以包括多个具有内部组件、各自是子系统的计算机设备。图42中所示的子系统经由系统总线75互连。绘示了额外的子系统,如打印机74、键盘78、存储装置79、与显卡82耦接的监视器76等。与I/O控制器71耦接的外围装置和输入/输出(I/O)装置可以通过所属领域中已知的任意数目个构件(如输入/输出(I/O)端口77(例如USB、))连接到计算机系统。举例来说,I/O端口77或外部接口81(例如以太网、Wi-Fi等)可以用于将计算机系统10连接到广域网,如因特网、鼠标输入装置或扫描仪。经由系统总线75的互连允许中央处理器73与每个子系统通信且控制来自系统存储器72或存储装置79(例如固定盘,如硬盘驱动器或光盘)的指令的执行以及子系统之间的信息交换。系统存储器72和/或存储装置79可以体现为计算机可读媒体。在此提及的任何数据可以从一个组件输出到另一个组件且可以输出到用户。计算机系统可以包括多个相同组件或子系统,例如通过外部接口81或通过内部接口连接在一起的多个相同组件或子系统。在一些实施例中,计算机系统、子系统或设备可以经由网络通信。在此类情况下,一个计算机可以视为客户端且另一个计算机视为服务器,其中每一者可以是同一计算机系统的一部分。客户端和服务器可以各自包括多个系统、子系统或组件。应了解,本发明的任何实施例可以按控制逻辑形式、以模块化或集成方式、使用硬件(例如专用集成电路或现场可编程门阵列)和/或使用通用可编程处理器的计算机软件来实施。如在此所用,处理器包括单核处理器、位于同一集成芯片上的多核处理器,或位于单个电路板上或网络化的多个处理单元。基于在此提供的揭露内容和教示内容,本领域的普通技术人员将知道并且了解使用硬件以及硬件与软件的组合来实施本发明的实施例的其它方式和/或方法。本申请中所述的任一种软件组件或函数可以作为软件代码实施,所述软件代码可通过使用任何适合计算机语言(如Java、C、C++、C#、面向对象的C语言、Swift,或脚本语言,如使用例如常规或面向对象技术的Perl或Python)的处理器执行。软件代码可以作为一系列指令或命令存储于计算机可读媒体上供存储和/或传输,适合的媒体包括随机存取存储器(RAM)、只读存储器(ROM)、磁性媒体(例如硬盘驱动器或软盘)或光学媒体(例如光盘(CD)或DVD(数字通用光盘))、闪存等。计算机可读媒体可以是此类存储或传输装置的任何组合。此类程序还可以使用载波信号来编码和传输,所述载波信号适合于经由符合多种协定的有线、光学和/或无线网络(包括因特网)传输。因此,根据本发明的一个实施例的计算机可读媒体可以使用以此类程序编码的数据信号产生。以程序代码编码的计算机可读媒体可以与兼容装置一起封装或与其它装置分开提供(例如经由因特网下载)。任何此类计算机可读媒体可以存在于单个计算机产品(例如硬盘驱动器、CD或整个计算机系统)上或内部,并且可以存在于系统或网络内的不同计算机产品上或内部。计算机系统可以包括监视器、打印机,或其它适合显示器以便将在此提及的任何结果提供给用户。在此所述的任何方法可以完全或部分地用计算机系统执行,所述计算机系统包括一或多个可经配置以执行所述步骤的处理器。因此,实施例可以涉及经配置以执行在此所述的任何方法步骤的计算机系统,其潜在地用不同组件执行相应步骤或相应的步骤群。本文中的方法步骤尽管作为编号的步骤呈现,但其可以同时或按不同顺序执行。另外,这些步骤的一部分可以结合其它方法的其它步骤的一部分使用。此外,步骤的全部或一部分可以是任选的。另外,任何方法的任何步骤都可以用执行这些步骤的模块、电路或其它构件来执行。特定实施例的具体细节可以按任何适合的方式组合而不脱离本发明实施例的精神和范围。然而,本发明的其它实施例可以涉及与每个个别方面或这些个别方面的特定组合相关的特定实施例。本发明的例示性实施例的以上描述已经为了说明和描述的目的而呈现。不希望其是穷尽性的或将本发明限制于所述的确切形式,并且根据以上教示内容,可以存在许多修改和变化。实施例是为了最佳地解释本发明的原理和其实际应用而选择和描述,由此使得本领域的其它技术人员能够在各种实施例中并且以适于所预期的特定用途的各种修改最佳地利用本发明。除非特别相反地指明,否则“一(a/an)”或“所述(the)”的叙述意指“一或多个”。除非特别相反地指明,否则“或”的使用意指“兼或”,而非“异或”。在此提及的所有专利、专利申请、公开和描述都整体并入供参考以用于于所有目的。不承认其是在先技术。附录A当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种新的多肽——dna甲基化酶13和编码这种多肽的多核苷酸的制作方法
- 具有治疗活性的环亚甲基-1,2-二羧酸酰胺,制备它们的方法和含它们的药物组合物的制作方法
- 组蛋白去乙酰化酶抑制剂的制作方法
- 基于dna聚合酶链反应的甲基化dna检测方法
- 检测Cre位点特异重组酶活性的转基因报告猪及培养方法
- 一种多肽——人含胞嘧啶特异性dna甲基化酶特征性序列片段的蛋白-9.35和编码这种 ...的制作方法
- 一种多肽——脱氧核糖核酸甲基化酶-9.35和编码这种多肽的多核苷酸的制作方法
- 利用酶活性的竞争性干扰和恢复检测被分析物的制作方法
- 一种基于化学发光反应的甲基化酶检测探针、检测试剂盒及检测方法
- 编码甲基化儿茶素生物合成酶的基因的制作方法