用于检测多种病原体的荧光实时检测方法及应用与流程
本发明属于核酸检测
技术领域:
,更具体地说,涉及用于检测多种病原体的荧光实时检测方法及应用。本发明还涉及采用该方法的定量和/或定性检测试剂盒。
背景技术:
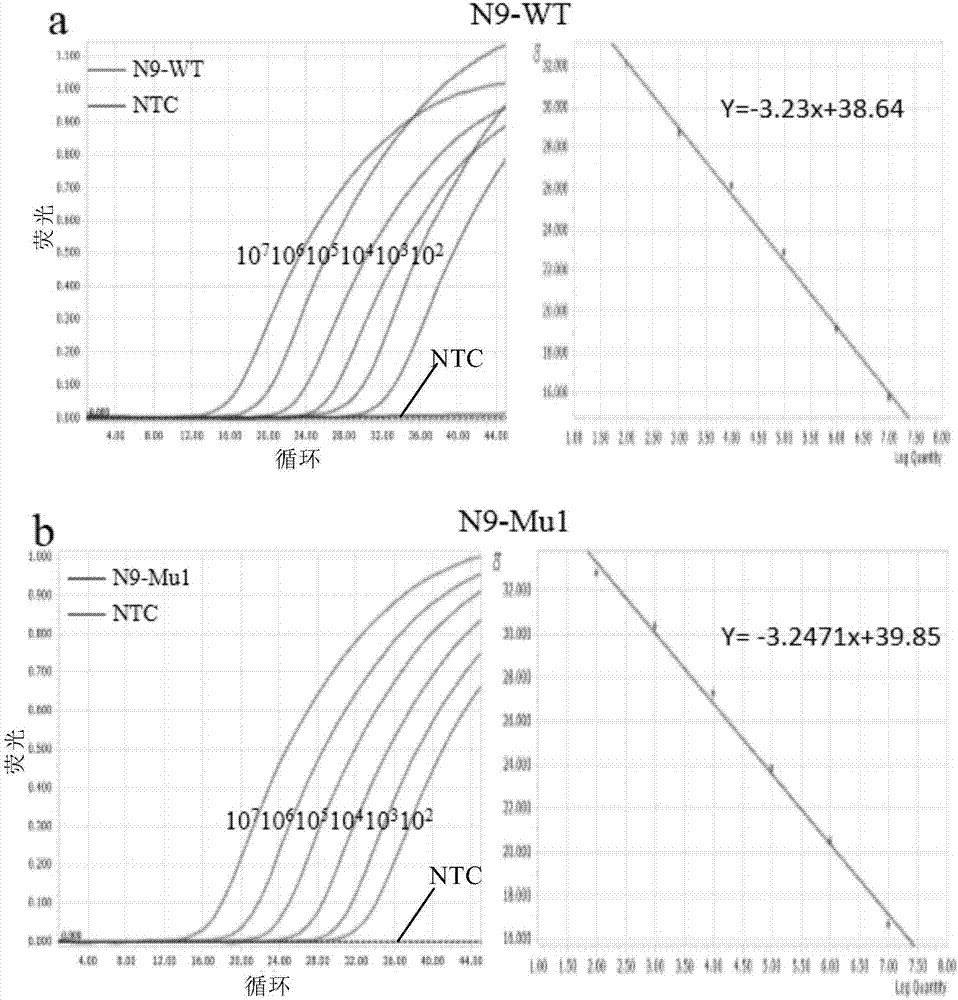
:病原检测是在疾病感染初期检测病原体,及时提供治疗,帮助预防疾病。病原体诊断一般分为细菌、病毒和真菌诊断。病毒检测有多重方法,包括病毒培养、电子显微镜观察、病毒抗原抗体检测。虽然病毒培养是检测的金标准,但是过程复杂,耗时数天。电子显微镜通过观察病毒的形态来进行科属判定,但是对技术人员要求高,且价格昂贵。病毒抗原抗体检测是检测病毒感染后产生的蛋白来鉴定病原体,还需要根据病人临床症状来判定。核酸检测是一种新型的检测方法,它通过pcr等手段检测病毒感染过程中所产生的核酸,不仅检测时间较早,且耗时较短。目前,实时荧光定量rt-pcr(real-timert-pcr)是较为常用的一种检测方法,分为染料法和探针法。然而,因病毒和细菌等微生物种类多,变异快,故易发生检测错误(如假阳性、假阴性)。为了检测多种类的病毒或同一种类病毒的不同病毒亚型或株,常常需要分别进行测试,因此增加了检测难度并费时费力。因此,本领域迫切需要开发简便、快速、灵敏、准确地检测高变异的病毒等病原体的方法。技术实现要素:本发明的目的就是提供简便、快速、灵敏、准确地检测高变异的病毒等病原体的方法。在本发明的第一方面,提供了一种定量或定性的核酸扩增方法,其特征在于,所述方法包括以下步骤:(i)在一扩增体系中,以待测核酸为模板,用m种引物对、高保真dna聚合酶进行核酸扩增,其中,m为≥1的正整数;所述的m种引物对中,至少一种引物对为位点特异性引物对,所述位点特异性引物对包括第一引物和第二引物;其中,(a)所述第一引物结构如下式i所示:z1-z2-z3-f1(i)式中,f1为荧光基团或淬灭基团;z1为无或附加功能区,长度为l1个核苷酸;z2为互补结合区,长度为l2个核苷酸;其中,z1和z2中至少一个与f2相连,其中f2为荧光基团或淬灭基团;并且f1和f2两者中一个为荧光基团,另一个为淬灭基团;z3为3’端的位点识别区,所述识别区的长度为l3个核苷酸,并且所述的识别区与待检测的模板检测区序列是匹配或非匹配的,并且l3为1-5;(b)所述第二引物为正常引物或与第一引物结构相同;(c)所述高保真dna聚合酶为具有外切酶活性的dna聚合酶;(ii)检测所述扩增体系中的荧光,从而获得定量或定性的检测结果。在另一优选例中,l1为0,或1-50个核苷酸。在另一优选例中,l2为10-50;较佳地12-40;更佳地15-35。在另一优选例中,l3为1、2、3,更佳地,1或2。在另一优选例中,第一引物和第二引物的长度为15-50个核苷酸;较佳地,20-40个核苷酸。在另一优选例中,所述m为≥2的正整数,较佳地,所述m为≥3的正整数,更佳地,所述m为≥4的正整数,更佳地,所述m为≥5的正整数。在另一优选例中,m为1-50、2-50、3-50、4-50或5-50。在另一优选例中,所述的m种引物对中≥80%,≥90%或100%是位点特异性引物对。在另一优选例中,所述正常引物是未经任何基团(包括荧光和淬灭基团)修饰的引物,其与模板序列完全互补配对。在另一优选例中,所述高保真dna聚合酶选自下组:q5dna聚合酶、kodplusneodna聚合酶、blendtaqdna聚合酶、promstarhsdna聚合酶、pfudna聚合酶、kodfxdna聚合酶、kodtmdna聚合酶、或其组合。在另一优选例中,所述高保真dna聚合酶选自下组:q5dna聚合酶或kodplusneodna聚合酶。在另一优选例中,所述荧光基团选自下组:fam、cy5、texasred、hex、vic、tet、joe、tamra、rox、lcred610、lcred640、lccyan500、yakimayellow、或其组合。在另一优选例中,所述封闭基团选自下组:bhq1、bhq3、eclipse、tamra、bhq2、dabcyl、或其组合。在另一优选例中,所述逆转录酶选自下组:m-mlvrtase、amvrtase、或其组合。在另一优选例中,所述的检测结果为检测多种病原体存在与否的检测结果。在另一优选例中,所述的多种病原体为3-100种,较佳地5-50种。在另一优选例中,所述的病原体包括同一种类病毒的不同病毒亚型或株。在另一优选例中,所述的病原体为高变异性病原体。在另一优选例中,其特征在于,所述扩增体系中还包括有少量的非高保真酶,所述非高保真dna聚合酶为不具有外切酶活性的dna聚合酶。在另一优选例中,所述非高保真dna聚合酶选自下组:taqdna聚合酶、tthdna聚合酶、或其组合。在另一优选例中,非高保真dna聚合酶的用量通常为0-1u/25ul,较佳地0.001-1u/25ul,更佳地0.01-0.2u/25ul,更佳地0.02-0.1u/25ul。在另一优选例中,非高保真dna聚合酶的用量与高保真dna聚合酶用量(按iu计)之比:1/20至1:1或更低,较佳地1/10-4/5,更佳地为1/5-3/5。在另一优选例中,高保真dna聚合酶的用量为能够进行有效扩增。在另一优选例中,高保真dna聚合酶的用量为0.3-1u/25ul,较佳地0.4-0.8u/25ul,更佳地0.5-0.7u/25ul。在另一优选例中,添加更可能低的非高保真dna聚合酶(如0.01-0.1u/25ul)。在另一优选例中,所述f1为荧光基团,f2为淬灭基团。在另一优选例中,在核酸扩增反应体系中,添加的mg2+离子终浓度为2mm-8mm,较佳地,3-7mm,更佳地,4mm-6mm。在另一优选例中,利用不同荧光和淬灭基团标记针对不同目标核酸的荧光引物,针对不同目标核酸进行单重或多重实时荧光核酸扩增。在另一优选例中,所述的方法是非诊断性和非治疗性的方法。在本发明的第二方面,提供了一种非诊断性和非治疗性地检测病原微生物的方法,其特征在于,所述方法包括以下步骤:(a)利用如本发明第一方面所述的核酸扩增方法,对待测样品进行实时荧光核酸扩增;和(b)分析实时荧光核酸扩增反应扩增曲线,从而判定所述待测样品是否存在病原微生物。在另一优选例中,检测结果可以通过荧光定量pcr检测反应中出现的荧光信号或扩增曲线,或者通过电泳检测扩增产物。在另一优选例中,通过标准曲线和扩增曲线的ct(阈循环数)值来确定基因的表达量,或者通过比较目的基因和参照基因的ct值进行相对定量。在本发明的第三方面,提供了如本发明的第一方面所述的核酸扩增方法的用途,其特征在于,用于体外检测病原微生物。在另一优选例中,用于对微生物核酸(真菌、细菌和病毒的rna、dna)和基因表达的mrna进行荧光实时定量检测。在另一优选例中,用于高变异性病原体的多重(定量)检测和人类致病相关基因的表达定量检测。在另一优选例中,所述的检测是生物安全或食品安全相关检测。应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。附图说明图1显示了用高保真dna聚合酶和荧光引物对核酸(耐受突变)进行检测原理图。图2显示了本发明一个实例中对h7n9病毒核酸进行检测的标准曲线。其中,a为检测野生型模板,b为检测突变型模板。图3显示了非高保真dna聚合酶用量对体系的影响。图4显示了本发明的检测体系可以有效检测各种突变位点。图5显示了本发明的荧光引物3’端标记荧光基团对扩增反应效率的影响。图6显示了不同高保真dna聚合酶对扩增反应效率的影响。图7显示了本发明方法可以进行多个荧光通道检测。其中,a、b、c分别表示使用5种高保真酶对fluc的探针(hex-bhq2)、actb的探针(texred-eclipse)、actb的探针(hex-bhq1)进行反应;d表示使用q5酶对piv4病毒探针(cy5-bhq3)进行反应;e、f表示使用7种高保真酶对rsva的探针(fam-mgb)和actb的探针(fam-tamra)进行反应。图8显示了不同长度目的产物对高保真dna聚合酶介导的荧光引物扩增体系的影响。图9显示了不同pcr反应添加剂对本发明方法的影响。其中,a为添加tamc,b为添加bt,c为添加ssb,d为添加甲酰胺,e为添加dmso。图10显示了本发明一个实例中对基因表达进行定量检测的结果。其中,a、b表示同时使用基于taqman探针的荧光方法和新方法对体外转录的β-actin基因rna标准品(107—102拷贝/μl)进行检测,并制作标准曲线;图10c表示以8份正常血液样本中抽提出的rna核酸为样本,使用普通荧光方法和新方法定量检测β-actin基因表达量。图11显示了不同mg2+浓度对扩增反应效率的影响。具体实施方式本发明人经过广泛而深入的研究,首次开发了一种可简便、快速、灵敏、准确地检测高变异的病毒等病原体的方法。具体地,本发明人利用荧光引物基于、任选的微量非高保真dna聚合酶、高保真dna聚合酶的3’-5’外切酶校正功能以及特定的优化条件,开发了一种操作简单快捷、灵敏性强、准确度高的检测方法。实验表明,本发明方法可广泛用于诊断和非诊断性的检测,并且可用于检测各种不同的病原体,如细菌、病毒等,尤其高变异性的病原体微生物。在此基础上完成了本发明。术语如本文所用,术语“本发明方法”和“本发明检测方法”可互换使用,指本发明第一方面中所述的基于高保真dna聚合酶、任选的微量非高保真dna聚合酶和荧光引物的、可简便、快速、灵敏、准确地检测高变异的病毒等病原体的方法。如本文所用,碱基的“匹配”或“配对”是指两条核苷酸序列中相应的碱基按照a与t,g与c配对的原则形成反向互补的双链结构。如本文所用,术语“完全匹配”是指引物与模板之间序列完全互补,不存在任何碱基错配。探针探针是一种寡核苷酸序列,与目标序列上游引物和下游引物之间的序列配对,探针5’端一般连接荧光基团,3’端连接淬灭基团。当完整的探针与目标序列配对时,荧光基团发射的荧光因与3’端的淬灭剂接近而被淬灭,但在进行延伸反应时,聚合酶的5’外切酶活性将探针进行酶切,使得荧光基团与淬灭剂分离。随着扩增循环数的增加,释放出来的荧光基团不断积累,因此荧光强度与扩增产物的数量呈正比关系,呈现一种s型曲线。本发明在这里将荧光引物命名为hfman探针,以区分于普通荧光定量中taqman探针。本发明可以对核酸进行有效定量和检测,其荧光探针不要求与模板错配,即荧光引物3’末端无论与模板错配与否,均可实现几乎等效的扩增。荧光引物对在本发明检测方法中,采用了结合于靶序列并有效进行扩增的引物对。在所述的引物对中,至少一个引物是特殊结构的荧光引物对。在本发明中,通常在引物3’端进行修饰或封闭。典型地,引物3’羟基被修饰和封闭的类型可以是荧光基团也可以是封闭基团。荧光引物可以与模板完全匹配,也允许荧光引物3’末端存在一个或一个以上与模板错配的任何碱基。荧光引物的荧光和淬灭基团可以分别标记在探针的5’或3’末端和3’或5’末端,标记在3’末端的淬灭或荧光基团起到封闭3’-oh的作用,使其在常规pcr扩增反应中无法延伸。代表性的荧光基团的例子包括(但并不限于):fam、cy5、texasred、hex、vic、tamra、tet、joe、rox、lcred610、lcred640、lccyan500、yakimayellow、或其组合。代表性的封闭基团的例子包括(但并不限于):bhq1、bhq3、tamra、eclipse、bhq2、dabcyl、或其组合。在本发明中,被修饰的核苷酸种类没有特别限制,可以是:a、t、c、g或其组合。在另一优选例中,采用在3’端标记荧光基团方式,这样可获得最佳的检测效果,并且可以进一步提高检测的灵敏度,更容易和更灵敏地反映高保真dna聚合酶对荧光引物的3’末端切割和延伸。在本发明中,优化的反应体系可以使用单荧光引物或多重荧光引物。高保真dna聚合酶在本发明中,一个重要的特征是采用高保真dna聚合酶。如本文所用,“高保真dna聚合酶”指具有识别发生错配的“引物-模板”复合物并从所述复合物中切除发生错配的引物的错配区(或错配碱基)的dna聚合酶。该术语优选识别和切除位于错配引物的3’端的错配碱基的dna聚合酶。在本发明中,所述的高保真dna聚合酶没有特别限制,可以包括任何具有3’-5’外切酶活性的dna聚合酶。代表性的例子包括(但并不限于):kodtmdna聚合酶、q5dna聚合酶、kodplusneodna聚合酶、blendtaqdna聚合酶、promstarhsdna聚合酶、pfudna聚合酶、kodfxdna聚合酶、或其组合,优选为,q5dna聚合酶或kodplusneodna聚合酶。非高保真dna聚合酶如本文所用,“非高保真dna聚合酶”具有5’端到3’端的dna聚合酶活性,但不能识别和切除位于错配引物的3’端的错配碱基的dna聚合酶。在本发明中,所述的非高保真dna聚合酶没有特别限制,可以包括任何不具有或基本上不具有3’-5’外切酶活性的dna聚合酶。代表性的例子包括(但并不限于):taqdna聚合酶、tthdna聚合酶、或其组合。本发明检测方法及其基本原理本发明提供了一种可用于快速、灵敏而准确地对病原体进行多重检测和/或基因表达定量检测的方法。本发明方法可以是诊断性或非诊断性的。为了便于理解,提供以下基本原理供参考。应理解,本发明的保护范围并不受所述基本原理的任何限制。本发明的一个主要原理如下:如图1所示。本发明方法基于高保真dna聚合酶、任选的微量非高保真dna聚合酶和荧光引物,利用高保真dna聚合酶的3’-5’外切酶活性识别和切除荧光引物3’端修饰的荧光或者淬灭基团。在本发明中,一方面,可将3’末端最后一个碱基(无论错配与否)进行切割,然后荧光基团和淬灭基团分离,从而使荧光引物释放出荧光;另一方面,暴露出荧光引物3’-oh,在高保真dna聚合酶和任选的微量非高保真dna聚合酶的作用下扩增延伸。扩增反应中,随着扩增产物的增加,荧光信号相应增加,从而实现对整个扩增反应的实时检测。无论模板与荧光引物3’末端是否匹配,都可以进行高效扩增。当模板与荧光引物3’末端不匹配时,高保真dna聚合酶同样能够将修饰识别为错配信号,切除荧光引物3’端最后一个核苷酸,继续进行dna合成;当模板与荧光引物3’末端完全匹配或者少数碱基错配时,由于引物被荧光基团或者淬灭基团所修饰以及体系中高保真酶含量高,导致高保真酶对基团进行识别,并且切除荧光引物的3’末端添加了淬灭基团或荧光基团标记的配对核苷酸,使得pcr可以正常进行。在本发明中,一种典型的基于高保真酶、任选的微量非高保真dna聚合酶和荧光引物的核酸扩增方法,包括步骤:步骤1,根据野生型基因序列设计正向荧光引物和反向普通引物,对其中正向荧光引物进行5’-,3’-末端的荧光基团和淬灭基团标记;步骤2,用所述正向荧光引物和反向普通引物对待测样品进行(rt-或实时荧光定量)pcr扩增反应;当引物与模板(突变型)完全互补配对时,高保真dna聚合酶对正向荧光引物进行3’-5’校对,封闭的3’-oh被成功暴露启动dna合成;当引物与模板(野生型)不完全匹配时,高保真dna聚合酶也将发挥其3’-5’校对功能,识别正向荧光引物的3’末端封闭基团为错配信号,利用其3’-5’外切酶活性,切除3’羟基被封闭的核苷,暴露出正常3’羟基,可以在聚合酶催化下形成3-5磷酸二酯键,从而进行dna合成(即pcr扩增)。由于体系中高保真酶活性很强,无论荧光引物3’末端与模板完全匹配还是有错配,高保真酶都将发挥3’-5’校准功能,将荧光或者淬灭基团进行识别和切割,进而产生高的扩增效率(原理示意图可参见图1)。步骤3,通过实时荧光(rt-)pcr扩增曲线的阈循环数比较或电泳检测扩增产物。在另一优选例中,主要通过荧光定量方法检测扩增曲线,比较阈循环数。在本发明中,除了采用高保真dna聚合酶之外,同时还可以添加或使用少量非高保真dna聚合酶,例如无3’-5’外切酶活性的普通耐热dna聚合酶,如taqdna聚合酶、tthdna聚合酶等。通常,非高保真dna聚合酶的用量与高保真dna聚合酶用量(按iu计)之比:1/20至1:1或更低,较佳地1/10-4/5,更佳地为1/5-3/5。在本发明中,非高保真dna聚合酶的用量通常为0-1u/25ul,较佳地0.001-1u/25ul,更佳地0.01-0.2u/25ul,更佳地0.02-0.1u/25ul。此外,在本发明中,高保真dna聚合酶的用量没有特别限制,只要能够进行有效扩增。典型的高保真dna聚合酶的用量为0.3-1u/25ul,较佳地0.4-0.8u/25ul,更佳地0.5-0.7u/25ul。例如,当非高保真dna聚合酶为taq酶,高保真dna聚合酶为q5聚合酶时,可以采用0-0.4u/25ultaq酶并采用0.5u/25ulq5聚合酶。在本发明中,优选添加更可能低的非高保真dna聚合酶(如0.01-0.1u/25ul),或者不添加非高保真dna聚合酶。在另一优选例中,当荧光引物3’末端与模板存在错配时,存在几个错配碱基的情况下,本发明的扩增效率不受影响。即荧光引物分别与完全互补配对的模板序列(野生型)和与其3’端最后一个或几个核苷酸存在错配碱基的模板序列(突变型)反应,检测荧光引物对野生型和突变型的扩增效率相似。另外,本发明还发现不同的高保真酶对于切割荧光引物封闭3’端的效率有所不同,其中以q5酶的效率最高。dmso及甲酰胺对本方法的灵敏度有一定的提高,而牛凝血酶、四甲基氯化铵等对本方法有抑制作用。当模板是dna时能够更加灵敏的检测出模板,而当模板是rna时,检测的灵敏度将会略低于dna模板。本发明适用于对高变异病原体的荧光实时定量检测。高变异性病毒存在多个亚型或基因型以及各种变异株,特异性引物设计困难。利用本发明进行高变异病原体(如病毒)检测时,将荧光引物的3’末端设计为与病毒基因组保守序列匹配或者不匹配的任何碱基,可以实现对病毒基因组的有效扩增。该检测方法可以允许荧光引物3’端与检测模板存在任何碱基的错配,容忍病毒基因组的高变异性,减少特异性引物设计难度,提高病毒检测广谱性,同时又不损害其检测的特异性。本发明的应用本发明可广泛用于诊断和非诊断性的检测,并且可用于检测各种不同的病原体,如细菌、真菌、病毒等,尤其高变异性的病原体微生物。在另一类优选例中,本发明的检测是针对食品、病原微生物(细菌、真菌、病毒等)的检测。此外,本发明还可以实现对微生物核酸(真菌、细菌和病毒的rna、dna)和基因表达的mrna进行荧光实时定量检测。此外,本发明可广泛用于传染病病原快速单重和多重检测及基因表达定量检测。本发明的主要优点(1)与现有所有应用于病原微生物核酸检测方法相比,在检测病原微生物(细菌、病毒等)的核酸检测过程中引物设计简单,检测操作简便快捷,检测结果准确可靠。(2)对于高变异病毒的检测能够更好的设计引物,解决了病毒高变异区域难以设计合适引物的问题,对于流行病的检测(特别是禽流感,hiv等)有非常大的便利性。下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如sambrook等人,分子克隆:实验室手册(newyork:coldspringharborlaboratorypress,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。实施例1、基于荧光引物和高保真dna聚合酶体系构建实时荧光rt-pcr检测h7n9流感病毒选取甲型流感病毒h7n9的ha片段作为rna模板,分别为野生型n9-wt和突变型n9-mu1序列。两条序列仅存在一个单核苷酸突变位点,突变型相对于野生型仅有一个a→g的点突变。设计正向荧光引物和反向普通引物,其中反向普通引物位于突变位点下游与相应位置野生型和突变型模板完全互补配对;正向荧光引物与野生型模板完全互补配对,且其3’末端位于单核苷酸突变位点处,即与突变型包含错配(如表1所示)。对正向荧光引物的5’末端可以进行fam修饰,3’末端可以进行bhq1修饰,反向普通正常引物无任何修饰,两条引物的tm值均为60℃左右。将体外转录的n9rna野生型和突变型从107拷贝/μl以10倍梯度依次稀释至102拷贝/μl,每个rt-pcr反应体系加3μl进行扩增,制作标准曲线,观察扩增效率。表1引物序列如下:正向荧光引物:5’-fam-atcatcaccgcccacagtgtacaa-bhq1-3’;反向正常引物:5’-acatcctggatttgccatca-3’(seqidno:4);反应体系如下:反应热循环条件如下:按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应,其中q5酶为neb公司产品(#m0493s),rtase为生工公司的amvreversetranscriptase(#b500999)。rt-pcr扩增曲线如图2所示。结果表明,扩增曲线伴随模板浓度的梯度变化也呈现梯度关系,利用本方法检测突变体和野生型的扩增效果都很好,动态线性范围均在107-102拷贝/μl。实施例2、非高保真dna聚合酶用量对体系的影响选取克隆自甲型流感的ha序列的质粒dna(野生型),对其进行体外转录成rna,以此作为实时荧光定量rt-pcr模板,使用与实施例1中完全相同的一个正向荧光引物和反向普通引物,在体系中加入大量的高保真酶,非高保真dna聚合酶加入不同的量,如0、0.2、0.5、1、1.2、1.5u/25μl的体系,并且加入无核酸酶的水最为阴性对照(ntc)。反应体系如下反应条件如实施例1。按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应,其中反应体系中的缓冲液,q5高保真酶均为neb公司产品,taqdna聚合酶为天根生物有限公司的产品(fp304)。反应体系中rna模板的量为104拷贝/μl。分析rt-pcr扩增曲线和ct值。结果如图3所示。根据阈循环数的比较,当taqdna聚合酶加入量为0-0.5u时,扩增曲线几乎一样,当taqdna聚合酶加入量逐渐增加时,会对扩增产生些许抑制。其中,添加少量taq酶或者不加入taq酶,加入0.5u的高保真酶(q5酶最优浓度)时,rt-pcr扩增曲线最好,扩增效率最好。实施例3、基于荧光引物与高保真dna聚合酶的检测体系对不同突变位点的耐受本发明选取人类基因β-actin(actb)进行pcr扩增和体外转录获得rna标准品(野生型actb-wt),并设计与其具有仅有单一核苷酸突变(a→g)的突变型模板序列(actb-mu1),仅有单一核苷酸突变(a→u)的突变型模板序列(actb-mu2),仅有单一核苷酸突变(a→c)的突变型模板序列(actb-mu3)。分别设计正向荧光引物和反向普通引物,其中反向普通引物位于突变位点下游且与相应位置野生型和突变型模板完全匹配;四种正向荧光引物3’末端则位于点突变处(如表2所示)。分别对正向荧光引物的5’末端进行hex荧光基团修饰,对3’端进行bhq1淬灭基团修饰,反向普通引物5’和3’末端均没有任何修饰。四种正向荧光引物actb-a-probe、actb-g-probe、actb-t-probe、actb-c-probe分别与四种模板actb-wt、actb-g、actb-u和actb-c完全配对。表2引物序列如下:正向荧光引物:5’-hex-tggacttcgagcaagagatggcca/g/t/c-bhq1-3’;反向正常引物:5’-attgccaatggtgatgacctg-3’(seqidno.19);反应体系和反应条件如实施例1。按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应,其中反应体系中的缓冲液,q5高保真酶均为neb公司产品,反应体系中rna模板的量为104拷贝。分析rt-pcr扩增曲线和ct值。结果如图4所示。根据阈循环数的比较,说明扩增各种突变模板的效率很相似,可以用于检测突变位点较多的病毒。另外,还研究了以dna作为初始反应模板进行同样的研究,结果与之相同。实施例4、正向荧光引物5’末端标记淬灭基团及3’末端标记荧光基团,对于高保真dna聚合酶识别切割及扩增反应效率的影响用克隆自甲型流感的ha序列的质粒dna(野生型和突变型)制备相应rna标准品。将所述rna标准品作为实时荧光定量rt-pcr模板,使用与实施例1中完全相同的一个正向荧光引物和反向普通引物序列,互换正向荧光引物5’-,3’-端修饰基团。此处将正向荧光引物的5’末端进行淬灭基团(bhq1)修饰,3’末端进行荧光基团fam修饰。将rna标准品从106拷贝/μl以10倍梯度依次稀释至104和103拷贝/μl,每个rt-pcr反应体系加1μl进行扩增,观察扩增反应效率。按照以下反应体系和程序进行反应。反应体系如下:反应热循环条件如实施例1。按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应。rt-pcr扩增曲线结果如图5所示。结果说明,3’端带有荧光基团修饰更有利于高保真dna聚合酶的识别与切割,也更有利于检测出不完全配对的突变型样品,有利于提高检测的准确性和灵敏度。实施例5、不同高保真dna聚合酶在本发明中的扩增反应效率差异用克隆自甲型流感的ha序列的质粒dna(野生型和突变型)制备相应rna标准品。将所述rna标准品作为实时荧光定量rt-pcr模板,使用与实施例1中完全相同的一个正向荧光引物和反向普通引物。分别用7种不同的高保真酶体系对n9-mu1模板进行扩增,这7种高保真酶分别为东洋纺(上海)生物科技有限公司的kodfx(kodfx-101),kodfxneo(kfx-201),kodplusneo(kod-401),和blendtaq-plus-(btq-201),北京康润诚业生物科技有限公司的expfudna聚合酶(a161-01,宝生物工程(大连)有限公司的primestarhsdna聚合酶和neb(北京)有限公司的热启动超保真2xmastermix(#m0494s)。rna标准品为106拷贝。反应体系如下:反应热循环条件如实施例1。除primestarhsdna聚合酶外,其他的六种dna聚合酶反应时均加入额外的3mmmgcl2。按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应,然后将扩增产物进行琼脂糖凝胶电泳鉴定,所用凝胶为2%琼脂糖凝胶,分子量标记为购自takara公司的50bpdnaladder(dyeplus)dnamarker(3421a)takaradl2,000dnamarker(d501a)。rt-pcr扩增曲线及产物电泳结果如图6所示,说明7种高保真dna聚合酶都可以利用荧光引物扩增突变型n9-mu1模板,扩增产物条带约84bp。通过对rt-pcr反应扩增曲线及阈循环数的比较发现,q5扩增时扩增曲线ct值最小,blendtaq,kodfxneo,kodplusneo和primstarhs的扩增曲线ct值几乎一样,ct值较高的是expfu和kodfx。实施例6、荧光引物两端不同荧光基团及淬灭基团修饰组合对于不同高保真dna聚合酶扩增效率的影响用克隆自人类基因β-actin(actb)制备相应rna标准品(野生型actb-wt),并设计与其具有仅有单一核苷酸突变(a→g)的突变型模板序列(actb-mu1)。分别设计正向荧光引物和反向普通引物,其中反向普通引物位于突变位点下游且与相应位置野生型和突变型模板完全匹配;正向荧光引物3’末端则位于点突变处(如表3所示)。分别对正向荧光引物的5’末端可以进行不同荧光基团修饰,3’末端可以进行相对的不同淬灭基团修饰(如表2所示),反向普通引物5’和3’末端均没有任何修饰。选用5种高保真dna聚合酶进行反应:东洋纺(上海)生物科技有限公司的kodplusneo(kod-401)和blendtaq-plus-(btq-201),北京康润诚业生物科技有限公司的expfudna聚合酶(a161-01),宝生物工程(大连)有限公司的primestarhsdna聚合酶和neb(北京)有限公司的热启动超保真2xmastermix(#m0494s);另外,本实施例还选取了丙型流感病毒(fluc),副流感病毒4型(piv4),呼吸道合胞病毒a型(rsva)特定片段作为rt-pcr反应模板,相应的设计了正向荧光引物和反向普通引物。分别对正向荧光引物进行不同荧光基团和淬灭基团的修饰。反应体系和程序如下:表3反向引物序列如下:actb反向正常引物r1:5’-attgccaatggtgatgacctg-3’(seqidno.19);piv4反向正常引物:5’-attctgatggttggaktctggtg-3’(seqidno.21);rsva反向正常引物:5’-gccaaggaagcatgcaataaa-3’(seqidno.22);fluc反向正常引物:5’-ttgccattcgtcccatctctc-3’(seqidno.23);反应体系如下:反应热循环条件如实施例1。按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应。rt-pcr扩增曲线如图7所示,表明5种高保真dna聚合酶都可以切割3’端bhq1,bhq3,eclipse,tamra封闭基团,但不能切割bhq2和mgb修饰基团延伸荧光引物。实施例7、不同长度目的产物对高保真dna聚合酶介导的荧光引物扩增体系的影响选择如实施例3中所述的rna标准品作为逆转录pcr反应模板rna,设计分别带5’fam修饰和3’tamra修饰的正向荧光引物,设计了两个反向普通引物,命名为r1和r2。两个反向普通引物分别与actb-mu1的下游序列完全互补配对。正向荧光引物与r1扩增出95bp产物,与r2扩增出140bp产物。表4seqidno.正向荧光引物序列tggacttcgagcaagagatggcca11反向正常引物序列r1attgccaatggtgatgacctg19反向正常引物序列r2caggaaggaaggctggaaga20反应体系如下:反应热循环条件如实施例1。按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应。rt-pcr扩增曲线(图8)表明,待扩增目的产物长度在90-150bp长度范围内对高保真dna聚合酶扩增效率没有明显影响。实施例8、不同pcr反应添加剂的使用对新方法的影响使用与实施例1中完全相同的突变型模板rna序列n9-mu1和反向普通引物。正向荧光引物序列3’末端修饰淬灭基团bhq1,5’末端修饰荧光基团fam。在体系中加入不同浓度不同种类的添加剂(甲酰胺(sigma)、二甲基亚砜(dmso,sigma)、四甲基氯化铵(tamc,sigma)、牛凝血酶(thermfisher)、单链结合蛋白(et-ssb,neb)),按照以下反应体系和程序进行反应。反应体系如下:反应热循环条件如实施例1。按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应。rt-pcr扩增曲线如图9所示。结果表明:加入以上添加剂,对rt-pcr的扩增促进作用都不强,有些甚至严重抑制其扩增。实施例9、对基因表达进行定量检测使用与实施例3中相同的荧光引物actb-g探针,对从血液中提取出的mrna进行β-actin基因表达定量检测。分别用本方法和市场上销售的普通荧光定量试剂盒(如天根一步法qrt-pcr试剂盒货号:fp304)检测β-actin基因表达进行比较。按照以下反应体系和程序进行反应:反应体系如下:反应热循环条件如实施例1。按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应。分析rt-pcr扩增曲线和8个样本检测结果如图10所示。结果表明,本方法可以很好的扩增rna,其检测效果明显好于天根试剂盒的检测效果,两种方法对8个样本β-actin基因表达定量检测无显著性差异,但是本方法检测的结果均比普通试剂盒检测值要高一些,表明本方法检测样本准确度更高,可以真实地反应实际情况。实施例10、体系中mgcl2浓度对反应的影响使用与实施例3中相同的荧光引物actb-a探针和actb-r1,分别作为体系的上下游引物,并且以体外转录的β-actin的rna为模板。在体系中额外添加终浓度为0、1、2、3和4mm的mgcl2,优化体系中mgcl2的浓度。按照以下反应体系和条件进行反应:反应体系如下:反应热循环条件如实施例1。按上述反应体系和反应条件在roche96实时荧光定量pcr仪上进行反应。分析rt-pcr扩增曲线(图11)可知,当体系中额外添加0-4mmmgcl2时,扩增曲线的峰值不断增高,扩增曲线的ct值均是一致的。由于体系中mgcl2浓度过高可能会导致反应的特异性降低,因此我们最终选取额外添加2-4mmmgcl2来增强反应。即体系中mg2+终浓度为4-6mm(提供的缓冲液中原本mgcl2浓度为2mm)。综上所述,根据高保真酶存在的3’-5’外切酶活性能够更丰富的设计特殊引物。因此可以基于荧光引物利用高保真酶和任选的微量非高保真酶对突变位点高或者低的微生物核酸进行定量检测以及基因表达定量检测。在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。序列表<110>中国科学院上海巴斯德研究所<120>用于检测多种病原体的荧光实时检测方法及应用<130>p2017-0539<160>23<170>patentinversion3.5<210>1<211>153<212>rna<213>甲型流感病毒<400>1agggaaacacucaaacggaacaauacacgauaggucccaguaucgcgcccugauaagcug60gccacuaucaucaccgcccacaguguacaacagcaggguggaaugcauuggguggucaag120uacuaguugccaugauggcaaauccaggauguc153<210>2<211>153<212>rna<213>突变型n9-mu1<400>2agggaaacacucaaacggaacaauacacgauaggucccaguaucgcgcccugauaagcug60gccacuaucaucaccgcccacaguguacagcagcaggguggaaugcauuggguggucaag120uacuaguugccaugauggcaaauccaggauguc153<210>3<211>24<212>dna<213>引物<400>3atcatcaccgcccacagtgtacaa24<210>4<211>20<212>dna<213>引物<400>4acatcctggatttgccatca20<210>5<211>184<212>rna<213>智人<400>5ggaaaucgugcgugacauuaaggagaagcugugcuacgucgcccuggacuucgagcaaga60gauggccacggcugcuuccagcuccucccuggagaagagcuacgagcugccugacggcca120ggucaucaccauuggcaaugagcgguuccgcugcccugaggcacucuuccagccuuccuu180ccug184<210>6<211>184<212>rna<213>突变型actb-mu1<400>6ggaaaucgugcgugacauuaaggagaagcugugcuacgucgcccuggacuucgagcaaga60gauggccgcggcugcuuccagcuccucccuggagaagagcuacgagcugccugacggcca120ggucaucaccauuggcaaugagcgguuccgcugcccugaggcacucuuccagccuuccuu180ccug184<210>7<211>184<212>rna<213>突变型actb-mu2<400>7ggaaaucgugcgugacauuaaggagaagcugugcuacgucgcccuggacuucgagcaaga60gauggccccggcugcuuccagcuccucccuggagaagagcuacgagcugccugacggcca120ggucaucaccauuggcaaugagcgguuccgcugcccugaggcacucuuccagccuuccuu180ccug184<210>8<211>184<212>rna<213>突变型actb-mu3<400>8ggaaaucgugcgugacauuaaggagaagcugugcuacgucgcccuggacuucgagcaaga60gauggccucggcugcuuccagcuccucccuggagaagagcuacgagcugccugacggcca120ggucaucaccauuggcaaugagcgguuccgcugcccugaggcacucuuccagccuuccuu180ccug184<210>9<211>24<212>dna<213>引物<400>9tggacttcgagcaagagatggcct24<210>10<211>24<212>dna<213>引物<400>10tggacttcgagcaagagatggccg24<210>11<211>24<212>dna<213>引物<400>11tggacttcgagcaagagatggcca24<210>12<211>24<212>dna<213>引物<400>12tggacttcgagcaagagatggccc24<210>13<211>27<212>dna<213>引物<400>13ttctgggatgagtggtgctctggctgt27<210>14<211>18<212>dna<213>引物<400>14tgctattgtgcactaaag18<210>15<211>26<212>dna<213>引物<400>15actggccttggagaagaagcactatc26<210>16<211>94<212>rna<213>副流感病毒4型<400>16agaucacaaugaacuacagaugccagaauuugaaaaugacaucaauccuuucaaucaucc60aagguucacagccagagcaccacucaucccagaa94<210>17<211>67<212>rna<213>呼吸道合胞病毒a型<400>17aauacagccaaaucuaaccaacuuuacacuacuacuucucaucaaauauccuuagugcac60aauagca67<210>18<211>157<212>rna<213>丙型流感病毒<400>18acuggccuuggagaagaagcacuaucucuccaaagacaaacagaaaguuuggccauauua60uguaaucacacuuuuggaaguaauauaaugagaccccacuuggaaaaagcaauaaaagga120guugaaggcagaguuggagagaugggacgaauggcaa157<210>19<211>21<212>dna<213>引物<400>19attgccaatggtgatgacctg21<210>20<211>20<212>dna<213>引物<400>20caggaaggaaggctggaaga20<210>21<211>23<212>dna<213>引物<400>21attctgatggttggaktctggtg23<210>22<211>21<212>dna<213>引物<400>22gccaaggaagcatgcaataaa21<210>23<211>21<212>dna<213>引物<400>23ttgccattcgtcccatctctc21当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种转基因鲑鱼AquAdvantage品系特异性实时荧光PCR检测引物、检测方法和试剂盒与流程
- 犬圆环病毒的实时荧光PCR检测用引物及检测方法与流程
- 用于荧光学图像的检测和实时显示的荧光学系统的制作方法与工艺
- 用于荧光学图像的检测和实时显示的荧光学系统的制作方法与工艺
- 利用单拷贝核基因检测食品和饲料中牛源性成分的实时荧光PCR检测方法与流程
- H10N7禽流感病毒的实时荧光PCR检测试剂盒及检测方法与流程
- 多重实时荧光PCR检测Cr(VI)还原复合菌群六种功能基因的方法与流程
- 刺苋的实时荧光PCR鉴定方法与流程
- 一种猪肺炎支原体的实时荧光PCR检测试剂盒及其用途的制作方法与工艺
- 实时荧光PCR特异性检测远志的引物及检测方法与流程
- 一种封闭式病原体检测卡盒的制作方法与工艺
- 检测16种呼吸道病原体的基因芯片检测用试剂盒及方法与流程
- 一种同时检测多种病原体的五重PCR检测方法与流程
- 逆转肿瘤多药耐药的基因组合物h‑R3/PAMAM G5/MDR1 siRNA及其应用的制作方法与工艺
- 用于对肉类、植物或植物部分进行病原体检测和病害治理的方法与流程
- 一种用于检测治疗恶性肿瘤的一线药物耐药基因表达量的试剂盒的制造方法与工艺
- 用于检测植物病原体的装置和方法与流程
- 用于检测多种病原体的荧光实时检测方法及应用与流程
- 一种用于检测手足口病相关病原体的LAMP引物组合物及其相关应用的制造方法与工艺
- 一种用于检测巴氏杆菌氯霉素类药物耐药基因的分子标记及其应用的制造方法与工艺