一种他唑巴坦酸、他唑巴坦二苯甲酯制备工艺与应用的制作方法
本发明涉及一种有机高分子合成工艺,特别涉及一种他唑巴坦酸、他唑巴坦二苯甲酯制备工艺与应用。
背景技术:
β-内酰胺类抗生素从诞生至今已有半个多世纪的历史。在此期间,随着细菌对常用抗生素不断产生耐药性,人们在不断的开发更高效且对耐药菌更有效的抗生素,同时针对耐药菌产生使β-内酰胺酶类失活的β-内酰胺酶这一机制,研制出与抗生素合用的β-内酰胺酶抑制剂,其中比较典型的就是他唑巴坦。他唑巴坦对各种类型的β-内酰胺酶甚至I型酶亦很有效,作用比舒巴克坦强10倍,稳定性比克拉维酸好,对多种质粒及染色体中介的β-内酰胺酶的抑制作用比克拉维酸和舒巴克坦强。与氨苄西林、阿莫西林等联用的体内外实验均取得了较满意的效果,具有毒性低,稳定性好,抑酶活性强等优点,曾在第三十届国际化疗会议上被评为最有前途的β-内酰胺酶抑制剂。
目前,较为常见的制备过程如下所示:
这种较为常规的制备工艺也是目前市面上合成的他唑巴坦酸/他唑巴坦钠的方式,但是由于中间的外加剂以及部分中间产物具有易燃易爆的特性或毒性较强,如乙炔、碘氧化钠等物质,造成现有技术中的生产工艺较为危险。
对比文件1:授权公告号为“CN102020663B”的中国专利所公开的一种他唑巴坦的合成方法;
对比文件2:申请公布号为“CN102382123A”的中国专利所公开的一种他唑巴坦钠的合成方法。
由于近年来,国际上对他唑巴坦的需求量越来越大,市场前景十分广阔,越来越多的实验室、公司开始对他唑巴坦进行研究;如对比文件1中采用新式的合成方法,以6-APA为原料,通过酯化、氧化、还原脱溴等连续反应不经分离制得关键中间体6,6-二氢青霉烷亚砜酸二苯甲酯,再与2-三苯基硅-1,2,3-三唑反应引入三唑环后,经高锰酸钾氧化、中间甲酚脱保护得最终产物他唑巴坦;虽然这种制备方式相对于现有技术而成,更为安全、环保,但是最终产率均值约在70%-80%之间,产率较低。
因而,在此我们提供一种环保且产率更好的制备工艺。
技术实现要素:
本发明的目的是提供一种产率较高且较为环保的他唑巴坦酸二苯甲酯的制备工艺。
本发明的上述技术目的是通过以下技术方案得以实现的:一种他唑巴坦酸二苯甲酯的制备工艺,合成步骤如下所示:
开环反应:
氯化反应:
缩合反应:
双氧化反应:
作为优选,包括以下步骤:
步骤1:开环反应
步骤1.1:在反应容器A中投入3.0-3.5份份甲苯、0.2-0.4份脱溴物和0.1-0.2份MBT,升温至70-75℃反应80-100分钟;
步骤1.2:反应完毕后,降温至58-75℃;
步骤1.3:将步骤1.2中产物减压浓缩至干,加入2.3-2.5份二氯甲烷搅拌至完全溶解得到澄清液体,得到开环物二氯甲烷溶液;
步骤2:氯化反应
步骤2.1:将步骤1.3中的开环物二氯甲烷溶液转入至反应容器B中持续搅拌并降温使得反应容器B中的开环物二氯甲烷溶液至0-5℃;
步骤2.2:向反应容器B中缓慢滴加浓度为31%的盐酸溶液0.3-0.5份,滴加温度小于0℃;
步骤2.3:再向反应容器B中滴加浓度为13-15%的亚硝酸钠溶液0.5-0.6份,控制滴加温度为5-10℃,滴加时间为85-100分钟;
步骤2.4:将反应容器B连通其中物质放置在5-10℃下保温10-12小时后,结束反应;
步骤2.5:离心机甩滤后,将滤液转入另一反应容器C中静置分层,水层用二氯甲烷提取,合并有机层后依次用水、碳酸氢钠水溶液、纯化水反复洗涤;
步骤2.6:将洗涤后的有机层转入反应容器D中,减压蒸馏至干,加入1.4-1.5份丙酮后得氯化物丙酮溶液备用;
步骤3:缩合反应
步骤3.1:将步骤2.6中制备得到的氯化物丙酮溶液转入反应容器E中中持续搅拌,再向其中加入0.4-0.5份纯化水和0.08-0.10份1,2,3-三氮唑,控制温度为25-30℃,控制时间为12-15小时;
步骤3.2:反应完毕后减压蒸馏至干;
步骤3.3:投入1.3-1.6份二氯甲烷并溶解得到澄清溶液,取有几层用纯化水洗涤干净后减压浓缩至干,得到糖浆物;
步骤3.4:投入0.3-0.4份乙酸乙酯后搅拌2.0小时至结晶后离心,控制搅拌温度为25℃,离心后甩干得到具有一定湿度的缩合物;
步骤4:双氧化反应
步骤4.1:将步骤3.4中制备得到的缩合物潮品和二氯甲烷投入反应容器F中搅拌得到澄清溶液;
步骤4.2:降温至10℃后,投入0.06-0.07份高锰酸钾后缓慢升温至30-35℃,保温3-4小时后结束反应;
步骤4.3:减压蒸馏至大量固体析出,再投入0.5-0.6份乙酸乙酯将固体打散,冷却至0-5℃后,静置1小时后离心,乙酸乙酯洗涤干净后,甩干。
作为优选,步骤1.1中采用HPLC监控,当脱溴物小于1.0%时停止步骤1.1;
步骤2.3中采用HPLC监控,当开环物小于0.5%时停止反应。
作为优选,步骤2.5中离心机甩滤之后,滤饼用0.2-0.25份的二氯甲苯洗涤,将滤液与有机层合并后,依次用水洗2-3次、1.5%的碳酸氢铵水溶液洗涤2-3次、纯化水洗涤。
本发明的目的是提供一种产率较高且较为环保的他唑巴坦酸的制备工艺。
本发明的上述技术目的是通过以下技术方案得以实现的:由上述方法制备得到的最终产物继续制备得到,合成步骤如下:
作为优选,包括以下步骤:
步骤5.1:向反应容器G中投入他唑巴坦二苯酯0.2-0.3份和0.5-0.9份间甲酚,升温至80℃后静置7小时;
步骤5.2:向反应容器H中投入1.0-1.2份乙酸乙酯,再将步骤5.1中得到的产物降温转入至反应容器H中,投入碳酸氢钠盐水、盐和纯化水搅拌10分钟后,静置分层;
步骤5.3:水相用乙酸乙酯洗涤后调节PH至4.0,加入EDTA、0活性炭脱色,脱色后压滤,再调节PH至2.0静置1小时后,再调节PH至1.5后保温离心得潮湿的他唑巴坦酸粗品;
步骤5.4:将0.13-0.15份纯化水、0.2-0.3份高浓乙醇均匀混合后升温至70℃后,将步骤5.3中制备得到他唑巴坦酸粗品加入后溶解;
步骤5.5:降温至0℃以下后,保温4小时后离心,用乙醇漂洗,烘干得他唑巴坦酸。
作为优选,采用浓度为10%的盐酸溶液调节PH。
本发明的目的是提供一种他唑巴坦酸二苯甲酯的应用。
本发明的上述技术目的是通过以下技术方案得以实现的:一种上述制备方法制备得到的他唑巴坦酸用于β-内酰胺酶抑制剂的制备。
本发明的目的是提供一种他唑巴坦酸二苯甲酯的应用。
本发明的上述技术目的是通过以下技术方案得以实现的:用于β-内酰胺抑制剂的制备。
综上所述,本发明具有以下有益效果:
(1)他唑巴坦酸作为青霉烷砜类β-内酰胺酶抑制剂,对I~VI型3-内酰胺酶均有效,特别是对I型酶更为有效;其毒性低,稳定性好,抑酶活性强;临床用于治疗多种细菌包括需氧菌和厌氧菌引起的感染,包括下呼吸道感染、皮肤和腹部感染等;而本方案中的制备方式在现有技术的碘代反应的基础上做出改进,引进氯代反应,优化反应质量,提高产品纯度,降低反应过程中有毒有害、易燃易爆物质的使用,使反应更加绿色、环保;
(2)以甲苯、脱溴物、2-巯基苯并噻唑、二氯甲烷为原料,经开环反应、氯代反应、双氧化反应等步反应后得到他唑巴坦酸,并能得到中间产物他唑巴坦二苯甲酯,以上两种物质均可作为产品销售,降低了生产成本,提高了销售利润;且开环反应、氯代反应、双氧化反应全程均使用HPLC监控,精准控制反应进程,提高产物纯净度;
(3)本方案中所提供的制备方法中不在使用乙炔、碘氧化钠等物质,而其中大量使用的甲苯、二氯甲苯、丙酮、乙酸乙酯、乙醇、间苯酚等物质均可循环使用,进一步的减少物料的浪费,符合环境友好化学;
(4)检测可得:采用本方案中制备得到的他唑巴坦酸和他唑巴坦二苯甲酯旋光度较好,且产率可高达98%-99.92%,制备成本也低于现有技术中的制备工艺。
附图说明
图1是实施例中他唑巴坦二苯甲酯的制备工艺流程图;
图2是实施例中他唑巴坦酸的制备工艺流程图。
具体实施方式
本具体实施例仅仅是对本发明的解释,其并不是对本发明的限制,本领域技术人员在阅读完本说明书后可以根据需要对本实施例做出没有创造性贡献的修改,但只要在本发明的权利要求范围内都受到专利法的保护。
实施例1:
他唑巴坦酸的制备工艺流程图,如图1所示,主要包括以下步骤:
步骤1:开环反应;
步骤2:氯化反应;
步骤3:缩合反应;
步骤4:双氧化反应。
开环反应的反应方程式如下所示:
制备步骤如下所示:
步骤1.1:在反应容器A中投入3.0份甲苯、0.2份脱溴物和0.1份MBT,升温至70℃反应80-100分钟;
步骤1.2:反应完毕后,降温至58℃;
步骤1.3:将步骤1.2中产物减压浓缩至干,加入2.3份二氯甲烷搅拌至完全溶解得到澄清液体,得到开环物二氯甲烷溶液。
氯化反应的反应方程式如下所示:
制备步骤如下所示:
步骤2.1:将步骤1.3中的开环物二氯甲烷溶液转入至反应容器B中持续搅拌并降温使得反应容器B中的开环物二氯甲烷溶液至0-5℃;
步骤2.2:向反应容器B中缓慢滴加浓度为31%的盐酸溶液0.3份,滴加温度小于0℃;
步骤2.3:再向反应容器B中滴加浓度为13%的亚硝酸钠溶液0.5份,控制滴加温度为5-10℃,滴加时间为85-100分钟;
步骤2.4:将反应容器B连通其中物质放置在5-10℃下保温10-12小时后,结束反应;
步骤2.5:离心机甩滤后,将滤液转入另一反应容器C中静置分层,水层用二氯甲烷提取,合并有机层后依次用水、碳酸氢钠水溶液、纯化水反复洗涤。
缩合反应的反应方程式如下所示:
制备步骤如下所示:
步骤3.1:将步骤2.6中制备得到的氯化物丙酮溶液转入反应容器E中中持续搅拌,再向其中加入0.4份纯化水和0.08份1,2,3-三氮唑,控制温度为25-30℃,控制时间为12-15小时;
步骤3.2:反应完毕后减压蒸馏至干;
步骤3.3:投入1.3份二氯甲烷并溶解得到澄清溶液,取有几层用纯化水洗涤干净后减压浓缩至干,得到糖浆物;
步骤3.4:投入0.3份乙酸乙酯后搅拌2.0小时至结晶后离心,控制搅拌温度为25℃,离心后甩干得到具有一定湿度的缩合物。
双氧化反应的反应方程式如下所示:
制备步骤如下所示:
步骤4.1:将步骤3.4中制备得到的缩合物潮品和二氯甲烷投入反应容器F中搅拌得到澄清溶液;
步骤4.2:降温至10℃后,投入0.06份高锰酸钾后缓慢升温至30℃,保温3-4小时后结束反应;
步骤4.3:减压蒸馏至大量固体析出,再投入0.5份乙酸乙酯将固体打散,冷却至0-5℃后,静置1小时后离心,乙酸乙酯洗涤干净后,甩干。
全称采用HPLC检测,在步骤1.1中,反应至60分钟以后,利用HPLC进行监测,当脱溴物小于1.0%时开始降温;步骤2.3中当开环物小于0.5%时停止反应。
实施例2:
步骤1:开环反应
步骤1.1:在反应容器A中投入3.5份份甲苯、0.4份脱溴物和0.2份MBT,升温至70-75℃反应80-100分钟;
步骤1.2:反应完毕后,降温至75℃;
步骤1.3:将步骤1.2中产物减压浓缩至干,加入2.5份二氯甲烷搅拌至完全溶解得到澄清液体,得到开环物二氯甲烷溶液;
步骤2:氯化反应
步骤2.1:将步骤1.3中的开环物二氯甲烷溶液转入至反应容器B中持续搅拌并降温使得反应容器B中的开环物二氯甲烷溶液至0-5℃;
步骤2.2:向反应容器B中缓慢滴加浓度为31%的盐酸溶液0.5份,滴加温度小于0℃;
步骤2.3:再向反应容器B中滴加浓度为13-15%的亚硝酸钠溶液0.6份,控制滴加温度为5-10℃,滴加时间为85-100分钟;
步骤2.4:将反应容器B连通其中物质放置在5-10℃下保温10-12小时后,结束反应;
步骤2.5:离心机甩滤后,将滤液转入另一反应容器C中静置分层,水层用二氯甲烷提取,合并有机层后依次用水、碳酸氢钠水溶液、纯化水反复洗涤;
步骤2.6:将洗涤后的有机层转入反应容器D中,减压蒸馏至干,加入1.5份丙酮后得氯化物丙酮溶液备用;
步骤3:缩合反应
步骤3.1:将步骤2.6中制备得到的氯化物丙酮溶液转入反应容器E中中持续搅拌,再向其中加入0.5份纯化水和0.10份1,2,3-三氮唑,控制温度为30℃,控制时间为12-15小时;
步骤3.2:反应完毕后减压蒸馏至干;
步骤3.3:投入1.6份二氯甲烷并溶解得到澄清溶液,取有几层用纯化水洗涤干净后减压浓缩至干,得到糖浆物;
步骤3.4:投入0.4份乙酸乙酯后搅拌2.0小时至结晶后离心,控制搅拌温度为25℃,离心后甩干得到具有一定湿度的缩合物;
步骤4:双氧化反应
步骤4.1:将步骤3.4中制备得到的缩合物潮品和二氯甲烷投入反应容器F中搅拌得到澄清溶液;
步骤4.2:降温至10℃后,投入0.07份高锰酸钾后缓慢升温至30-35℃,保温3-4小时后结束反应;
步骤4.3:减压蒸馏至大量固体析出,再投入0.6份乙酸乙酯将固体打散,冷却至0-5℃后,静置1小时后离心,乙酸乙酯洗涤干净后,甩干。
全称采用HPLC检测,在步骤1.1中,反应至60分钟以后,利用HPLC进行监测,当脱溴物小于1.0%时开始降温;步骤2.3中当开环物小于0.5%时停止反应。
实施例3:
步骤1:开环反应
步骤1.1:在反应容器A中投入3.2份甲苯、0.25份脱溴物和0.14份MBT,升温至70-75℃反应80-100分钟;
步骤1.2:反应完毕后,降温至65℃;
步骤1.3:将步骤1.2中产物减压浓缩至干,加入2.37份二氯甲烷搅拌至完全溶解得到澄清液体,得到开环物二氯甲烷溶液;
步骤2:氯化反应
步骤2.1:将步骤1.3中的开环物二氯甲烷溶液转入至反应容器B中持续搅拌并降温使得反应容器B中的开环物二氯甲烷溶液至0-5℃;
步骤2.2:向反应容器B中缓慢滴加浓度为31%的盐酸溶液0.37份,滴加温度小于0℃;
步骤2.3:再向反应容器B中滴加浓度为13-15%的亚硝酸钠溶液0.53份,控制滴加温度为5-10℃,滴加时间为85-100分钟;
步骤2.4:将反应容器B连通其中物质放置在5-10℃下保温10-12小时后,结束反应;
步骤2.5:离心机甩滤后,将滤液转入另一反应容器C中静置分层,水层用二氯甲烷提取,合并有机层后依次用水、碳酸氢钠水溶液、纯化水反复洗涤;
步骤2.6:将洗涤后的有机层转入反应容器D中,减压蒸馏至干,加入1.4-1.5份丙酮后得氯化物丙酮溶液备用;
步骤3:缩合反应
步骤3.1:将步骤2.6中制备得到的氯化物丙酮溶液转入容器E中中持续搅拌,再向其中加入0.43份纯化水和0.089份1,2,3-三氮唑,控制温度为27℃,控制时间为13小时;
步骤3.2:反应完毕后减压蒸馏至干;
步骤3.3:投入1.4份二氯甲烷并溶解得到澄清溶液,取有几层用纯化水洗涤干净后减压浓缩至干,得到糖浆物;
步骤3.4:投入0.36份乙酸乙酯后搅拌2.0小时至结晶后离心,控制搅拌温度为25℃,离心后甩干得到具有一定湿度的缩合物;
步骤4:双氧化反应
步骤4.1:将步骤3.4中制备得到的缩合物潮品和二氯甲烷投入容器F中搅拌得到澄清溶液;
步骤4.2:降温至10℃后,投入0.065份高锰酸钾后缓慢升温至32℃,保温3-4小时后结束反应;
步骤4.3:减压蒸馏至大量固体析出,再投入0.53份乙酸乙酯将固体打散,冷却至0-5℃后,静置1小时后离心,乙酸乙酯洗涤干净后,甩干。
全称采用HPLC检测,在步骤1.1中,反应至60分钟以后,利用HPLC进行监测,当脱溴物小于1.0%时开始降温;步骤2.3中当开环物小于0.5%时停止反应。
实施例4:
步骤1:开环反应
步骤1.1:在反应容器A中投入3.4份份甲苯、0.35份脱溴物和0.18份MBT,升温至75℃反应80-100分钟;
步骤1.2:反应完毕后,降温至70℃;
步骤1.3:将步骤1.2中产物减压浓缩至干,加入2.3-2.5份二氯甲烷搅拌至完全溶解得到澄清液体,得到开环物二氯甲烷溶液;
步骤2:氯化反应
步骤2.1:将步骤1.3中的开环物二氯甲烷溶液转入至反应容器B中持续搅拌并降温使得反应容器B中的开环物二氯甲烷溶液至0-5℃;
步骤2.2:向反应容器B中缓慢滴加浓度为31%的盐酸溶液0.45份,滴加温度小于0℃;
步骤2.3:再向反应容器B中滴加浓度为13-15%的亚硝酸钠溶液0.58份,控制滴加温度为5-10℃,滴加时间为85-100分钟;
步骤2.4:将反应容器B连通其中物质放置在5-10℃下保温10-12小时后,结束反应;
步骤2.5:离心机甩滤后,将滤液转入另一反应容器C中静置分层,水层用二氯甲烷提取,合并有机层后依次用水、碳酸氢钠水溶液、纯化水反复洗涤;
步骤2.6:将洗涤后的有机层转入反应容器D中,减压蒸馏至干,加入1.4-1.5份丙酮后得氯化物丙酮溶液备用;
步骤3:缩合反应
步骤3.1:将步骤2.6中制备得到的氯化物丙酮溶液转入反应容器E中中持续搅拌,再向其中加入0.48份纯化水和0.09份1,2,3-三氮唑,控制温度为28℃,控制时间为12-15小时;
步骤3.2:反应完毕后减压蒸馏至干;
步骤3.3:投入1.5份二氯甲烷并溶解得到澄清溶液,取有几层用纯化水洗涤干净后减压浓缩至干,得到糖浆物;
步骤3.4:投入0.36份乙酸乙酯后搅拌2.0小时至结晶后离心,控制搅拌温度为25℃,离心后甩干得到具有一定湿度的缩合物;
步骤4:双氧化反应
步骤4.1:将步骤3.4中制备得到的缩合物潮品和二氯甲烷投入反应容器F中搅拌得到澄清溶液;
步骤4.2:降温至10℃后,投入0.068份高锰酸钾后缓慢升温至32℃,保温3-4小时后结束反应;
步骤4.3:减压蒸馏至大量固体析出,再投入0.56份乙酸乙酯将固体打散,冷却至0-5℃后,静置1小时后离心,乙酸乙酯洗涤干净后,甩干。
全称采用HPLC检测,在步骤1.1中,反应至60分钟以后,利用HPLC进行监测,当脱溴物小于1.0%时开始降温;步骤2.3中当开环物小于0.5%时停止反应。
实施例5:
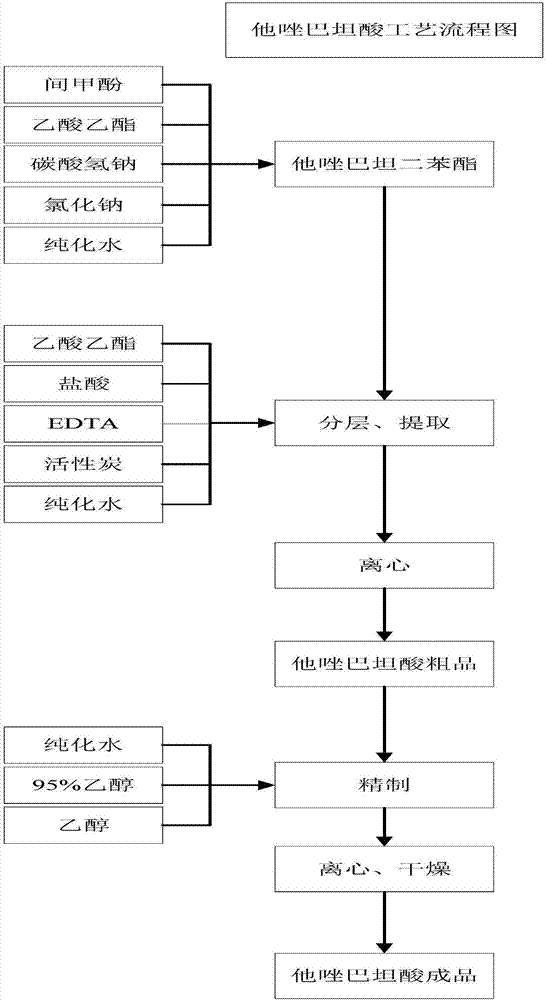
由实施例1-实施例4中制备得到的最终产物他唑巴坦二苯甲酯继续制备他唑巴坦酸,如图2所示,反应方程式如下所示:
制备步骤如下所示:
步骤5.1:向反应容器G中投入他唑巴坦二苯酯0.2份和0.5份间甲酚,升温至80℃后静置7小时;
步骤5.2:向反应容器H中投入1.0份乙酸乙酯,再将步骤5.1中得到的产物降温转入至反应容器H中,投入碳酸氢钠盐水、盐和纯化水搅拌10分钟后,静置分层;
步骤5.3:水相用乙酸乙酯洗涤后调节PH至4.0,加入EDTA、0活性炭脱色,脱色后压滤,再调节PH至2.0静置1小时后,再调节PH至1.5后保温离心得潮湿的他唑巴坦酸粗品,采用浓度为10%的盐酸调节PH;
步骤5.4:将0.13份纯化水、0.2份高浓乙醇均匀混合后升温至70℃后,将步骤5.3中制备得到他唑巴坦酸粗品加入后溶解;
步骤5.5:降温至0℃以下后,保温4小时后离心,用乙醇漂洗,烘干得他唑巴坦酸。
实施例6:
步骤5.1:向反应容器G中投入他唑巴坦二苯酯0.3份和0.9份间甲酚,升温至80℃后静置7小时;
步骤5.2:向反应容器H中投入1.2份乙酸乙酯,再将步骤5.1中得到的产物降温转入至反应容器H中,投入碳酸氢钠盐水、盐和纯化水搅拌10分钟后,静置分层;
步骤5.3:水相用乙酸乙酯洗涤后调节PH至4.0,加入EDTA、0活性炭脱色,脱色后压滤,再调节PH至2.0静置1小时后,再调节PH至1.5后保温离心得潮湿的他唑巴坦酸粗品,采用浓度为10%的盐酸调节PH;
步骤5.4:将0.15份纯化水、0.3份高浓乙醇均匀混合后升温至70℃后,将步骤5.3中制备得到他唑巴坦酸粗品加入后溶解;
步骤5.5:降温至0℃以下后,保温4小时后离心,用乙醇漂洗,烘干得他唑巴坦酸。
实施例7:
步骤5.1:向反应容器G中投入他唑巴坦二苯酯0.23份和0.7份间甲酚,升温至80℃后静置7小时;
步骤5.2:向反应容器H中投入1.1份乙酸乙酯,再将步骤5.1中得到的产物降温转入至反应容器H中,投入碳酸氢钠盐水、盐和纯化水搅拌10分钟后,静置分层;
步骤5.3:水相用乙酸乙酯洗涤后调节PH至4.0,加入EDTA、0活性炭脱色,脱色后压滤,再调节PH至2.0静置1小时后,再调节PH至1.5后保温离心得潮湿的他唑巴坦酸粗品;
步骤5.4:将0.14份纯化水、0.25份高浓乙醇均匀混合后升温至70℃后,将步骤5.3中制备得到他唑巴坦酸粗品加入后溶解;
步骤5.5:降温至0℃以下后,保温4小时后离心,用乙醇漂洗,烘干得他唑巴坦酸。
实施例8:
步骤5.1:向反应容器G中投入他唑巴坦二苯酯0.28份和0.8份间甲酚,升温至80℃后静置7小时;
步骤5.2:向反应容器H中投入1.1份乙酸乙酯,再将步骤5.1中得到的产物降温转入至反应容器H中,投入碳酸氢钠盐水、盐和纯化水搅拌10分钟后,静置分层;
步骤5.3:水相用乙酸乙酯洗涤后调节PH至4.0,加入EDTA、0活性炭脱色,脱色后压滤,再调节PH至2.0静置1小时后,再调节PH至1.5后保温离心得潮湿的他唑巴坦酸粗品,采用10%的盐酸进行调节;
步骤5.4:将0.14份纯化水、0.28份高浓乙醇均匀混合后升温至70℃后,将步骤5.3中制备得到他唑巴坦酸粗品加入后溶解;
步骤5.5:降温至0℃以下后,保温4小时后离心,用乙醇漂洗,烘干得他唑巴坦酸。
测量实施例1-实施例8中制备得到的最终产物他唑巴坦二苯甲酯、他唑巴坦酸的纯净度并记录在表1中,以下测量结果均为多次测量后所取平均值。
表1:
将实施例1-实施例4中制备得到的最终产物带入进实施例5-实施例8中制备成他唑巴坦酸,并对制备得到的他唑巴坦酸进行测量,将测量标准及结果取平均值后记录在表2中。
表2:
- 还没有人留言评论。精彩留言会获得点赞!