一种配对互作基因检测方法及预测模型与流程
本发明涉及基因芯片技术,特别是一种配对互作基因检测方法及预测模型。
背景技术:
基因芯片技术也称dna微阵列(microarray),是一种从转录组水平检测基因表达情况的高通量技术。基因芯片技术的出现,使得从大规模基因表达水平上探究复杂疾病机理成为可能。基因选择是芯片数据分析的重要任务,基因选择即从上万个表达基因中筛选出与肿瘤表型相关的信息基因。芯片技术能一次检测上万个基因的表达量,但并不是每个基因都与肿瘤表型相关,过多的无关、冗余基因可导致过拟合、维数灾等问题,且不利于肿瘤发病机制研究。通过基因选择剔除无关基因,选择一个相关子集至少有以下三个优点:1)提高分类(诊断)模型的泛化性;2)基于少量的基因实施高精度诊断,可以大幅度降低诊断成本,是实现临床应用的基础;3)选择与肿瘤表型相关的基因有利于进一步实现肿瘤机理解析。
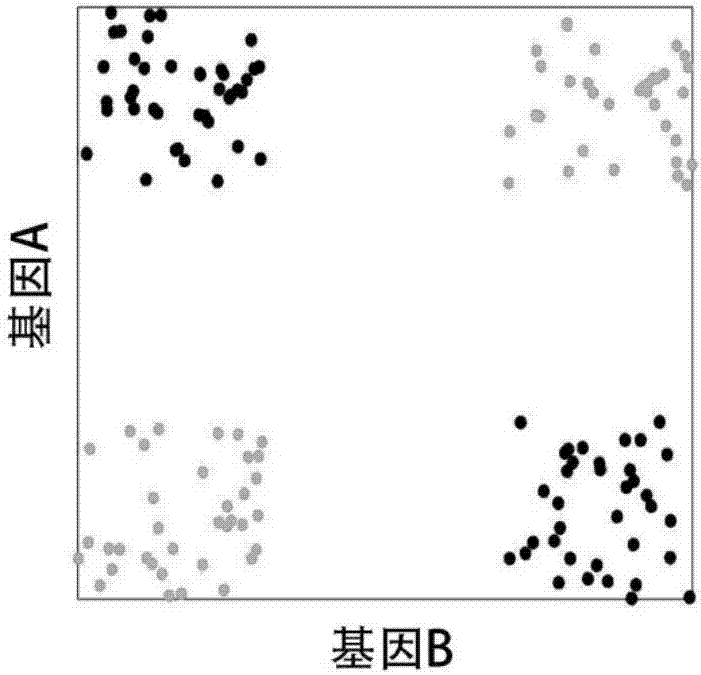
信息基因选择对临床诊断、药物靶点筛选以及疾病机理研究等意义重大。大量研究表明,基于多基因的功能通道变异而非单基因变异是导致复杂疾病的主要原因。传统单基因过滤方法(例如t测验[1])仅能筛选与疾病相关的单效应基因,无法检测到与疾病相关的互作基因。两两基因配对互作是基因互作的最基本形式,现今已有大量研究致力于筛选与复杂疾病关联的配对互作基因,图1-图4展示了典型的配对互作基因情形:单独基于基因a(genea)无法有效区分得病样本与健康样本,单独基于基因b(geneb)同样无法有效区分得病样本与健康样本,当同时考虑genea与geneb时可有效区分得病样本与健康样本。配对互作基因检测方法可分为两大类:第一类为基于互信息的非转换策略,如基于系统树的方法(dendrogram-basedmethod)[2]与三变量最大信息系数法(mic(x1;x2;y))[3]。对基因的连续表达值离散化是互信息方法面临的主要问题,基于系统树的方法采用系统进化树方法进行离散化,但易导致信息丢失,并不能检测到典型的配对互作基因;三变量最大信息系数法基于三维寻优方法计算三变量互信息,可检测配对互作基因,但属于计算密集型算法,计算效率过高,并且其检测到的互作基因并不能直接提高预测模型精度。第二类为转换策略,如doublets[4]。对基因p与基因q,doublets方法引入一个新的变量z代替两个初始基因表达值,共有四种转换模式:
sum模式:zis=xip+xiq(1)
diff模式:zis=xip–xiq(2)
mul模式:zis=xip×xiq(3)
sign模式:
将包含n个样本,m个基因的基因表达数据定义为{yi,xij},i=1,2,…,n;j=1,2,…,m。xij为第i个样本的第j个基因的表达值,yi表示第i个样本的疾病表型(可用1表示不得病,2表示得病)。经表达值转化后,计算新变量z与y之间的相关性。该方法具有计算效率高的优点,但不能有效检测典型的配对互作基因。
技术实现要素:
本发明旨在提供一种计算效率高的配对互作基因检测方法及预测模型,有效提高检测精度。
为解决上述技术问题,本发明所采用的技术方案是:一种配对互作基因检测方法,包括以下步骤:
1)对给定的数据集{yi,xij},首先对每个基因表达值进行秩规格化(即对于任一基因j,将其表达值按从小到大顺序排列,然后用每个表达值的排列位置代替初始表达值),得到规格化后的数据矩阵{yi,rij};
2)对任意两个基因p和基因q采用abs模式进行转换,获得转换后的数据集{yi,zis};abs模式:zis=|rip–riq|;其中i=1,2,…,n;p=1,2,…,m;q=1,2,…,m;p≠q;s=1,2,…,m(m-1)/2;n为数据集中的样本数,m为数据集中的基因数;rip表示第i个样本的第p个基因排秩转换后的表达值;riq表示第i个样本的第q个基因排秩转换后的表达值;
3)基于t测验方法计算每个变量zis与表型yi之间的t值,根据t值对应的概率值p值判断两个基因之间的互作效应。
本发明中,对应p值小于0.05表示某一对基因互作达到显著水平;p值小于0.01表示某一对基因互作达到极显著水平。
相应地,本发明还提供了一种配对互作基因预测模型,包括分类器,且该分类器的输入变量为经abs模式转换后得到:第i个样本的第p个基因排秩转换后的表达值rip和第i个样本的第q个基因排秩转换后的表达值riq;其中,abs模式:zis=|rip–riq|;其中i=1,2,…,n;p=1,2,…,m;q=1,2,…,m;p≠q;s=1,2,…,m(m-1)/2;n为数据集中的样本数,m为数据集中的基因数;rip表示第i个样本的第p个基因排秩转换后的表达值;riq表示第i个样本的第q个基因排秩转换后的表达值。
与现有技术相比,本发明所具有的有益效果为:本发明基于abs模式转换,可有效检测到与疾病关联的配对互作基因,并且因为在重要性评价时,采用的是简单的t测验方法,具有计算效率高的优点。另外,以经过abs模式转换后的新的变量可有效提高模型预测精度。
附图说明
图1-图4表示典型的配对互作基因模式(黑色为病例样本,灰色为健康样本);
图5abs模式筛选的最强互作基因对;
图6mic(x1;x2;y)方法筛选的最强互作基因对;
图7sum模式筛选的最强互作基因对;
图8diff模式筛选的最强互作基因对;
图9mul模式筛选的最强互作基因对;
图10sign模式筛选的最强互作基因对;
图11dendrogram-based方法筛选的最强互作基因对。
具体实施方式
本发明基于转换策略提出了一种新的配对互作基因快速检测方法。对给定的数据集{yi,xij},但基因芯片数据存在噪音,假设有500个样本,对某个基因而言,其它样本的表达值可能都是200左右,但有一个样本的表达值达到1000,所以首先对每个基因表达值进行秩规格化[5],得到规格化后的数据矩阵{yi,rij}。例如对基因j,将其表达值按从小到大顺序排列,然后用每个表达值的排列位置代替初始表达值。然后对任意两两基因(假设为基因p与基因q)采用如下所示的abs模式进行转换:
abs模式:zis=|rip–riq|(5)
式(5)中,i=1,2,…,n;p=1,2,…,m;q=1,2,…,m;p≠q;s=1,2,…,m(m-1)/2。此时可获得转换后的数据集{yi,zis}。基于t测验方法可以计算每个变量z与表型y之间的t值。若对应的概率值(p值)小于0.05则表示z与y之间的关联显著,且对应t值越大,表示z与y之间的关联越强,同样表示对应的两个基因之间具有强的互作效应。p值是在做t测验时基于给定自由度下的t分布自动计算出来,规定p值小于0.05为显著水平,表示配对基因有互作效应的概率为95%,若p值小于0.01表示极显著水平,指配对基因有互作效应的概率为99%。基于t值排序,可获得具有强互作效应的配对互作基因。本发明发现,在构建预测模型时,若将互作基因的初始表达值(即xp与xq)作为分类器的输入变量,并不能获得较好的预测精度。本发明提出,在构建预测模型时,需将经abs模式转换后的变量(zs)作为分类器输入变量,可有效提高模型的预测精度。假设,检测到k对配对互作基因,不转换时有2×k个变量作为模型输入,转换策略有k个变量作为模型输入。
将本发明的方法应用于4个真实基因芯片数据,其样本大小、基因个数以及数据来源见表1。
表1四个二分类基因表达数据
首先分别以doublets的四个模式、基于系统树的方法(dendrogram-basedmethod)、三变量最大信息系数法(mic(x1;x2;y))以及本发明方法abs模式检测prostate1数据中的配对互作基因。选择互作效应最强的一对基因,结果如图5-11所示,仅abs模式(图5)与mic(x1;x2;y)方法(图6)可以有效检测到如图3所示的典型配对互作基因。doublets的四个模式(图7-10)所检测的基因虽然能区分得病样本与健康样本,但其并非图1-4所示的典型配对互作基因,而属于典型单效应基因。
表2给出了上述不同样本数时几种方法的计算效率。当样本数较小(200),mic(x1;x2;y)方法完成一对互作计算耗时0.009秒,dendrogram-basedmethod需要0.66秒,而基于转换的方法(doublets以及abs模式法)仅耗时约0.0002秒。随着样本数增加,非转换方法耗时急剧增加,当5000样本时,mic(x1;x2;y)方法完成一对互作计算耗时16秒,dendrogram-basedmethod需要929秒,而基于转换的方法(doublets以及abs模式法)仅耗时约0.06秒。显然,基于转换的方法具有较高的计算效率。
表2不同方法的计算效率比较(单位:秒)
进一步以lung、prostate2、cardiovascular三个数据验证abs所检测的配对互作基因的预测性能。分类器选用支持向量分类(supportvectorclassification,svc),基于libsvm[10]平台实现,核函数为径向基核,惩罚参数c(c∈[2-5,215])与核函数参数γ(γ∈[2-15,23])采用grid.py程序寻优获得。预测性能评价指标为5次交叉测试精度acc(accuracy):将数据随机划分为5等份,首先以其中4份作为训练集,预测另外一份;重复5次,则可得到所有样本的预测结果。acc为样本判对率。单效应基因采用t测验方法排序,即计算每个基因与表型y间的t值,按降序排列。互作基因由abs模式方法选择得到。全部5次交叉测试acc见表3。基于前5、10、20对互作基因,当以初始基因表达值作为svc输入变量,三个数据的平均精度分别为72.10%、75.18%、78.67%,当将配对的基因表达值经abs转换后再作为svc输入变量,三个数据的平均精度分别提高到了75.58%、81.67%、84.63%。转换后的互作基因预测精度与前10、20、40个单效应基因的预测结果(77.30%、78.74%、80.36%)相当。最后,组合前10个单效应基因与5对互作基因(共20个基因),其平均预测精度为83.74%,明显高于20个单效应基因的平均预测精度(78.74%);同样,组合前20个单效应基因与10对互作基因(共40个基因),其平均预测精度为85.75%,明显高于40个单效应基因的平均预测精度(80.36%)。以上结果表明:1)直接以配对互作基因作为分类器输入向量,不能提高模型预测精度,需要进行abs转换;2)经abs转换后,互作基因预测性能与单效应基因预测性能相当;3)abs转换后互作基因能提高单效应基因的预测性能。
表3不同输入特征下的5次交叉测试精度(%)
注:“top10_单效应”表示由t测验获得的最强的前10个单效应基因;“top5_转换互作对”表示由abs模式方法获得的最强的前5对互作基因,并以abs转换后的5个新变量作为svc输入向量;“top5_非转换互作对”表示由abs模式方法获得的最强的前5对互作基因,以对应的初始10个基因表达值作为svc输入向量。
参考文献
[1]jafarip,azuajef.anassessmentofrecentlypublishedgeneexpressiondataanalyses:reportingexperimentaldesignandstatisticalfactors[j].bmcmedicalinformaticsanddecisionmaking,2006,6(1):27.
[2]watkinsonj,wangx,tianz,anastassioud.identificationofgeneinteractionsassociatedwithdiseasefromgeneexpressiondatausingsynergynetworks[j].bmcsystemsbiology,2008,2(1):10.
[3]cheny,caod,gaoj,yuanzmdiscoveringpair-wisesynergiesinmicroarraydata[j].scientificreports,2016,6(6):30672.
[4]choprap,leej,kangj,lees.improvingcancerclassificationaccuracyusinggenepairs[j].plosone,5(12):e14305.
[5]gemand,d'avignonc,naimandq,winslowrl.classifyinggeneexpressionprofilesfrompairwisemrnacomparisons[j].statisticalapplicationsingenetics&molecularbiology,2004,3(1):article19.
[6]singhd,febbopg,rossk,jacksondg,manolaj,laddc,etal.geneexpressioncorrelatesofclinicalprostatecancerbehavior[j].cancercell,2002,1(2):203-209.
[7]spiraa,beaneje,shahv,steilingk,liug,schembrif.etal.airwayepithelialgeneexpressioninthediagnosticevaluationofsmokerswithsuspectlungcancer[j].naturemedicine,2007,13(3):361-366.
[8]penneykl,sinnottja,tyekuchevas,gerket,shuiim,kraftp,etal.associationofprostatecancerriskvariantswithgeneexpressioninnormalandtumortissue[j].cancerepidemiology,biomarkers&prevention,2015,24(1):255-260.
[9]ellsworthdl,jrcd,weyandtj,sturtzla,blackburnhl,burkea,etal.intensivecardiovascularriskreductioninducessustainablechangesinexpressionofgenesandpathwaysimportanttovascularfunction[j].circulation-cardiovasculargenetics,2014,7(2):151-160.
[10]changcc,lincj.libsvm:alibraryforsupportvectormachines[j].acmtransactionsonintelligentsystemsandtechnology,2011,2(3):27-52.
- 还没有人留言评论。精彩留言会获得点赞!