基于Y染色体分子标记高效推断姓氏的方法与流程
本发明涉及分子生物学、法医学和生物信息技术领域,具体地说,涉及一种基于y染色体分子标记高效推断姓氏的方法。
背景技术:
基因dna分子标记的遗传分析技术给法医侦查带来了革命性的变化。近30年来,dna分析已经成为法医调查的必备手段,各级公安机关都建立了大量相关人群的遗传数据库。其中,y染色体dna数据库,特别是微卫星(y-str)数据库是建立最早、规模最大、人口覆盖最广的遗传数据库。近年来,随着芯片技术和新一代测序技术的发展,y染色体snp数据也在不断积累。虽然,这些数据库及其相应数据的分子标记在亲子鉴定和身份识别方面发挥了重要作用,但是,目前对y染色体dna众多分子标记分析结果的信息利用仍非常局限,没有充分发挥y染色体dna大数据库的应用潜能。
y染色体绝大部分dna,即非重组区(non-combiningregionofy,nry),遵从严格的父系遗传,即只通过父亲遗传给男性后代。这种遗传模式与我国传统的姓氏继承模式非常相似,即绝大多数新生男丁出生后都随父亲的姓氏。由于y染色体dna的进化很大程度上与姓氏的传演相互平行,二者在演化上具有高度相关性,从理论角度,根据男性的y染色体dna上的变异模式可以对其姓氏进行推测。同时,中国自古以来就有寻根问祖的传统,对宗脉有着强烈的认同感。从北宋《百家姓》到现在由国务院人口普查办公室统计列出的中国名义上最新姓氏人口数目排名来看,姓氏的组成基本没有变化,其稳定性可有效提升姓氏推测的可靠性和准确性。这类推断结果可以直接应用于法医学分析,指导刑侦调查,尽可能的缩小调查范围,提高侦查效率。然而,目前尚没有根据y染色体dna的信息对中国姓氏进行推断的现成方法。因此,发展基于y染色体dna分子标记推断姓氏的分析方法非常必要。
此外,基于y染色体dna分子标记推断姓氏的分析方法还具有广泛的社会人文应用前景。
技术实现要素:
本发明的目的是提供一种基于y染色体dna分子标记高效推断姓氏的方法。
为了实现本发明目的,本发明提供y染色体分子标记在推断姓氏中的应用。
本发明还提供基于y染色体分子标记高效推断姓氏的方法,基于y染色体分子标记,利用统计学方法计算待测样本与数据库样本之间的最小遗传距离,从而得到待测样本的候选姓氏。
其中,所述分子标记包括str、snp或rflp等常用的,且位于y染色体非重组区域的分子遗传标记。
本发明进一步提供基于y染色体dna分子标记高效推断姓氏的方法,以y-str标记为例包括以下步骤:
步骤1:提取待测样本y的基因组dna,设计引物或利用商品化试剂盒对样本y染色体dna位点进行基因型分析;
步骤2:根据步骤1中相应位点的数据信息,包括基因型、等位基因或短片段重复数量等,计算待测样本y与数据库样本x之间的最小遗传距离d(y,x),定义待测样本与各已知姓氏样本之间的最小遗传距离ds=minx∈sd(y,x),其中sm={x:x姓氏为m};
遗传距离ds可根据所使用的y染色体dna的数据信息类型进行相应定义;具体地,针对y-str数据,可采用以下两种方法计算ds:
(1)余弦距离(cosinedistance,dcos)
其中,xi和yj分别表示样本x和y的str位点i和j上的短片段重复数量;
(2)溯祖距离(coalescencedistance)
其中,t为样本x和y的溯祖时间,ne为有效群体大小,μ=2.5×10-3为str位点的平均突变速率,n为分析中应用到的str位点的总数,
给定μ、n、k和ne两样本溯祖时间为t的概率可表示为:
其中,
步骤3:根据ds对姓氏按升序排列;
步骤4:选择前c位姓氏作为待测样本y的候选姓氏;其中,1≤c≤数据库中姓氏总数目。
前述的方法,步骤1中使用的试剂盒可以是ampf
前述的方法,步骤1所述的y-str位点包括但不限于dys19(dys394)、dys388、dys389、dys390、dys391、dys392、dys393、dys393(dys395)、dys413、dys425/dyf371、dys426、dys434、dys435、dys436、dys437、dys438、dys439(y-gata-a4)、dys441、dys442、dys443、dys444、dys445、dys446、dys447、dys448、dys449、dys450、dys452、dys453、dys454、dys455、dys456、dys458、dys459a&b、dys460(y-gata-a7.1)、dys461(y-gata-a7.2)、dys462、dys463、dys464、dys481、dys485、dys487、dys490、dys494、dys495、dys497、dys504、dys505、dys508、dys518、dys520、dys522、dys525、dys531、dys532、dys533、dys534、dys540、dys549、dys556、dys557、dys565、dys570、dys572、dys573、dys575、dys576、dys578、dys589、dys590、dys594、dys607、dys612、dys614、dys626、dys627、dys632、dys635(y-gata-c4)、dys636、dys638、dys641、dys643、dys710、dys714、dys716、dys717、dys724(cdy)、dys725、dys726、dyf385s1、dyf387s1a/b、dyf397、dyf399、dyf401、dyf406s1、dyf408、dyf411、dxys156、ycaiia&b、y-gata-h4、y-gata-a10、y-ggaat-1b07。
优选地,步骤1所述的y-str位点为dys19、dys389i、dys389ii、dys390、dys391、dys392、dys393、dys437、dys438、dys439、dys448、dys456、dys458、dys635和ygatah4。
前述的方法,步骤4中c为1-n之间的整数,其中n为预定的目标姓氏数目。
前述的方法,当步骤2中数据库样本量≥50000时,采用余弦距离或溯祖距离计算ds,优选余弦距离,以增加推断时效;当步骤2中样本量<50000时,采用余弦距离或溯祖距离计算ds,优选溯祖距离,以提高准确性。
本发明还提供一种基于y染色体dna分子标记技术开发的用于推断姓氏的装置,所述装置包括计算待测样本与数据库样本之间的最小遗传距离的模块及数据分析模块。
其中,计算模块是根据待测样本y的y染色体dna位点信息,计算待测样本y与数据库样本x之间的最小遗传距离d(y,x),定义待测样本与各已知姓氏样本之间的最小遗传距离ds3minx∈sd(y,x),其中sm={x:x姓氏为m}。
遗传距离d(y,x)的计算模块可包含但不限于下述两个子模块,其中一个子模块用于计算余弦距离,另一个子模块用于计算溯祖距离。
(1)余弦距离(cosinedistance,dcos)
其中,xi和yj分别表示样本x和y的str位点i和j上的短片段重复数量;
(2)溯祖距离(coalescencedistance)
其中,t为样本x和y的溯祖时间,ne为有效群体大小,μ=2.5×10-3为str位点的平均突变速率,n为分析中应用到的str位点的总数,
给定μ、n、k和ne两样本溯祖时间为t的概率可表示为:
其中,
数据分析模块用于分析待测样本y与数据库中候选姓氏之间的最小遗传距离。
借由上述技术方案,本发明至少具有下列优点及有益效果:
本发明提供的方法可根据y染色体分子标记,特别是y-str遗传变异信息对中国人群的姓氏进行有效推断,且姓氏推断的准确性随着每个姓氏样本量的增加而提高,随使用的y染色体遗传标记的数目增多而上升。采用本方法可以对中国人群的姓氏进行准确可靠的推断,并具有广阔的实际应用前景。
附图说明
图1为本发明较佳实施例中分析样本姓氏频率分布。
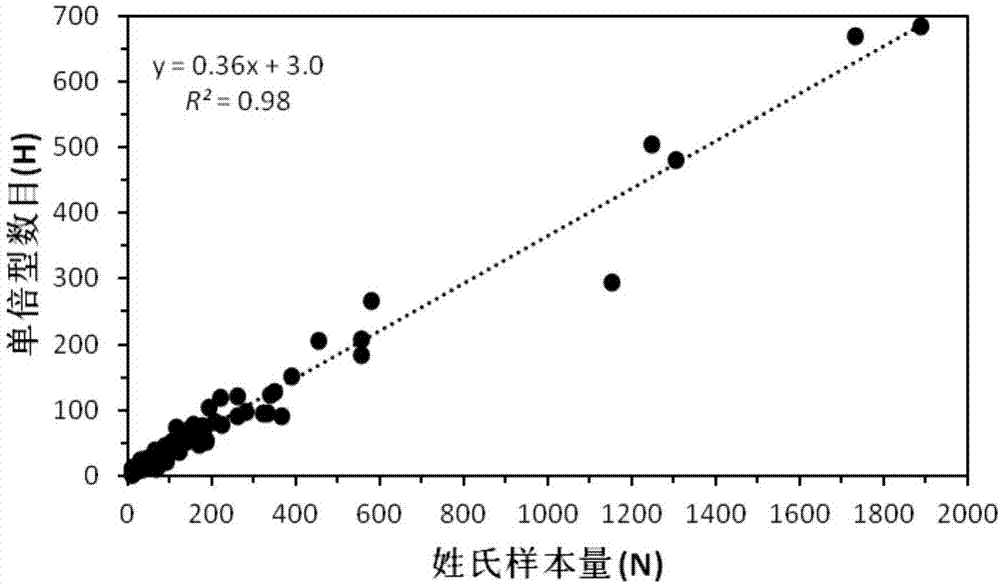
图2为本发明较佳实施例中姓氏样本量与其包含的单倍型之间的关系。
图3为本发明较佳实施例中姓氏推断准确性及其与候选姓氏数目之间的关系。
图4为本发明较佳实施例中姓氏样本量与姓氏推断准确性之间的关系。
图5为本发明较佳实施例中姓氏推断准确性与y-str位点数目之间的关系。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段,所用原料均为市售商品。
实施例基于y-str分子标记高效推断姓氏的方法
1.样本采集与基因型分析
19009位男性的血样由山东省公安局于2012-2014年实施的y-str数据库项目建立过程中采集,其中包含266个姓氏。样本中99.6%的个体为山东籍居民,0.4%的个体来自山东省以外的19个省份。各姓氏的样本量从1位到1889位不等,平均值为71位。有5个姓氏样本量超过1000,38个姓氏的样本量超过100;超过一半(130)的姓氏样本量不足10,其中46个姓氏仅有1个样品。样本姓氏的频率分布与我国人口姓氏分布相似(图1)。
基因组dna根据chelex-100的说明提取;提取dna的质量用
2.算法
假设数据库中包含了充足的已知姓氏和y-str变异谱的样本,y-str变异谱可以用向量x=(x1,x2,...,xn)来表示,其中第i个元素对应y-str位点i的等位基因大小。对于一个已知y-str变异谱的样本y=(y1,y2,...,yn),通过以下3个步骤搜索数据库并记录c(1-10)个姓氏作为候选姓氏:
步骤1:根据y-str数据,计算待测样本y与数据库样本x之间的最小遗传距离d(y,x),定义待测样本与各已知姓氏样本之间的最小遗传距离ds=minx∈sd(y,x),其中sm={x:x姓氏为m};
采用以下两种方法计算ds:
(1)余弦距离(cosinedistance,dcos)
其中,xi和yj分别表示样本x和y的str位点i和j上的短片段重复数量;
(2)溯祖距离(coalescencedistance)
其中,t为样本x和y的溯祖时间,ne为有效群体大小,μ=2.5×10-3为str位点的平均突变速率,n为分析中应用到的str位点的总数,
给定μ、n、k和ne两样本溯祖时间为t的概率可表示为:
其中,
步骤2:根据ds对姓氏按升序排列;
步骤3:选择前c位姓氏作为待测样本y的候选姓氏(c=1…c,c<数据库中姓氏总数目)。
3.数据分析
(1)数据预处理与质量控制:在分型的17个y-str位点中,dys385ab包含两个重复的str位点,无法明确具体等位基因的基因型,dys447在很多个体中数据缺失;剔除这两个y-str位点后,最终有15个y-str位点用于数据分析(姓氏推断)。数据格式如表1所示。
(2)缺失数据插补:对个别几个位点少数缺失数据进行插补(imputation),形成分析输入数据。
(3)姓氏推断:运用上述15个y-str位点,根据dcos和dcoal距离进行姓氏推断。结果的准确率通过5折交叉验证来评估。具体操作如下:首先将整个数据库分为5个大致相等的子集,1个子集作为验证集,其他4个作为测试集(类似y-str数据库)。通过将5个子集中每一个作为验证集来进行交叉验证。姓氏推断的平均准确率通过计算所有5个测试集中准确匹配的比例来确定。给定c个候选姓氏,准确匹配是指待测的姓氏属于推断获得的候选姓氏集。我们将c值设置为1到10进行一系列推断分析。
此外,我们从15个y-str中随机挑选3、6、9、12个y-str位点进行上述分析,以研究y-str遗传信息量对姓氏推断的影响。
4.准确性评价和结果评估
4.1数据特征
分析数据包括19,009名男性,含266个姓氏。姓氏的样本量从1位到1889位不等,平均值为71位。根据样本量排序,前100个姓氏呈指数分布(图1)。大约有10%的个体在一个及以上y-str位点数据缺失。不考虑缺失数据,在用于姓氏推断的15个y-str的等位基因数目从5(dsy437)到12(dsy389ii)个不等,平均(±sd)8.33±1.88(n=17,077)个。整个样本的基因多样性为0.9986±0.0001。较常见的126个姓氏(样本量≥10)人口占整个样本人口的97.6%;其基因多样性介于0.2857至1之间(表2)。姓氏频率等级与y-str基因多样性显著相关(spearman’srankcorrelationrho=0.46,p=5.93×10-8);单倍型数目与姓氏样本量线性相关(r2=0.98),表明常见姓氏的父系遗传多样性更高(图2)。
4.2姓氏推断准确性
基于最小遗传距离dcoal和dcos推断姓氏的性能如图3所示。总体而言,dcoal对姓氏推断的准确性略高于dcos,但当只指定1个候选姓氏时例外,dcoal的准确性比dcos低。总体而言,姓氏推断的准确性随指定候选姓氏数目的增加而提高。当用dcoal时,整体准确率介于65.21%(c=1candidate)到86.44%(c=10candidates)之间;当用dcos时,整体准确率在65.38%~86.02%(图3)。当指定候选姓氏的数目超过4(c≥4)时,两种距离对姓氏推断的准确率均>80%。但姓氏样本量较小时(如<10)推断的误差较大;排除这些样本,姓氏推断的准确率整体提高1%。
虽然dcoal对姓氏推断的准确性略高于dcos,但其运算强度远比dcos高。当c=10时,dcoal耗时100秒,而dcos只用2秒;即dcos比dcoal快50倍。
4.3姓氏样本量对推断准确性的影响
每个姓氏所包含的个体数目对姓氏推断具有明显影响,分析姓氏的样本量越大,姓氏推断的变异越小,分析结果越稳定。以余弦距离(dcos)为例,当推断姓氏的样本量小于10时,推断结果准确性的误差很大。但当量大于1000时,误差显著减小。此外,候选姓氏越多,准确性的误差越小(图4)。
4.4y-str信息量对姓氏推断准确性的影响
通过从15个y-str位点中随机抽取3、6、9、12个位点分别对姓氏推断来评估,相关结果见图5。不管指定候选姓氏的数目如何变化(c=1~10),姓氏推断的准确性都随y-str的增加而升高。当指定候选姓氏超过8个时,用9个y-str位点推断的准确性就与用15个y-str位点的结果趋近,但用15个位点的准确性还是显著优于用12个位点推断的结果。以上结果表明,随着y-str位点数目的增加,姓氏推断的准确性明显升高。
以上实施例表明本发明方法可以用于y-str对中国人群的姓氏进行有效推断。同时,姓氏推断的准确性随着每个姓氏样本量的增加而提高,随使用的y-str数目增多而上升。我国有近14亿人口,包含7000多个姓氏,因此可以推断中国姓氏的实际样本量远远比本实例涉及的样本量要大,故可以确定仅用实施例中涉及的15个y-str即可对我国人群的姓氏进行可靠推断。此外,实施例中最多仅用了15个y-str基因型数据,当前我国公共安全采集的遗传数据涉及的y-str位点通常超过17个,因此,可以断定根据当前收集的y-str数据对中国人群姓氏推断的准确性会更高。综上,本方法可对中国人群的姓氏进行可靠准确推断并具有广阔的实际应用前景。
采用本发明提供的方法可根据当前法医遗传学分析常用的15个y-str位点对我国人群的姓氏进行有效推断;在一定条件下推断的准确性超过80%(图3和图4),表明本方法可以有效地利用人群姓氏和y-str的数据信息。虽然,本方法有待于在更大的人群中进行验证,但是,本次测试数据中姓氏等级的频率分布(thezipfplot)(图1),大致反映了中国人口姓氏频率分布的特点(baeketal.2007)。因此,可以推断本方法在整个中国人群中也会有效。由此认为y-str数据是中国人群姓氏追踪中很有应用前景的数据来源,将有助于未来的法医调查工作。
以上研究结果进一步明确了姓氏的样本量对姓氏推断的准确性有重要影响。姓氏推断的准确性随姓氏样本量的升高呈现明显上升趋势(图4)。当姓氏样本量较小时推断的准确性的变异较大,但当样本量足够大时,推断的准确性趋于饱和。例如,当姓氏样本量从223到1899变化时,准确率在82%~93%之间浮动。这一现象指导我们在实际应用中,应将遗传分析的精力多投入到样本量较小,或者说罕见姓氏上。
虽然用更多的y-str位点会获得更准确的姓氏推断结果,但我们的研究结果表明agcuy18str荧光检测试剂盒中的15个y-str位点可对中国人群的姓氏进行有效推断。当用12个y-str位点时,姓氏推断的准确率趋于饱和(图5)。但需说明,这只是本分析案例中的一个现象,尚需进一步研究。
以上分析案例中姓氏推断高度的准确性表明中国人群的姓氏与父系遗传谱系之间存在高度的关联性。中国人群的以下几个特征可能对这种高度关联性相关:首先,中国人的姓氏具有很长的演化历史。欧洲的姓氏起源于中世纪,而中国的姓氏起源历史可追溯到4000年前。在中国姓氏较长的演化时间里y-str有望积累更多的遗传突变,据此我们可以区分不同的姓氏。
其次,由于文化传统的约束使得中国姓氏具有长期的保守型、稳定性和连续性的特点,这进一步增加了姓氏和y-str遗传的同步性(或共祖性)。这种社会文化特征可能提高姓氏和y-str平行传演的概率。这一点也反映在姓氏群体大小和y-str多样性的线性关系上(图1和图2,表2)。由于中国姓氏较长的历史,因此中国姓氏更有可能反映了人群更深层次的遗传分化,所有更容易被检测到。
最后,中国姓氏的分布呈现突出的地理特点。虽然一些大姓氏目前呈现全国分布,但是,大多姓氏在自然社区(自然村、镇、县市)多呈现聚集分布。这一点在我们分析的数据中也有反映(图1)。因此,一个小地方同姓的个体更有可能起源于相同的祖宗。这一现象自然会导致姓氏的分离与y-str分化程度相一致。
姓氏与y染色体同步分离为我们根据y-str推断姓氏提供了良好的契机。虽然这类方法可能会很有效,但也存在一些局限性。比如私生子、领养、改姓等这些情况可导致姓氏与y-str变异不一致,无法根据y-str对姓氏进行推断。需要指出,我们分析样本对姓氏的地理覆盖范围有限,因此,我们的方法有待进一步检验。同时,我们分析样本对中国姓氏的代表性尚不充分,仅包含266个姓氏,而且我们的结果主要来自对样本量超过10人的126个姓氏的分析,因此仅代表了中国姓氏和遗传多样性的一小部分。今后将用更大的数据对本发明方法进行检验。
尽管如此,从初步分析结果来看,本方法在姓氏推断中具有良好的应用前景。对于区域性的数据,我们的方法可以根据少数几个y-str位点对姓氏精细准确推断。历史早期的姓氏变迁,将对姓氏推断造成挑战,但是可以通过把大数据分割为若干较小的区域性数据集,对其分别进行姓氏推断,予以解决。这一问题也可以通过在姓氏推断过程中利用更多的地理、社会文化信息来解决。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之做一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!