一种克里唑替尼中间体及其制备方法和应用与流程
本发明涉及克里唑替尼
技术领域:
,具体为一种克里唑替尼中间体的盐及其制备方法和用途。
背景技术:
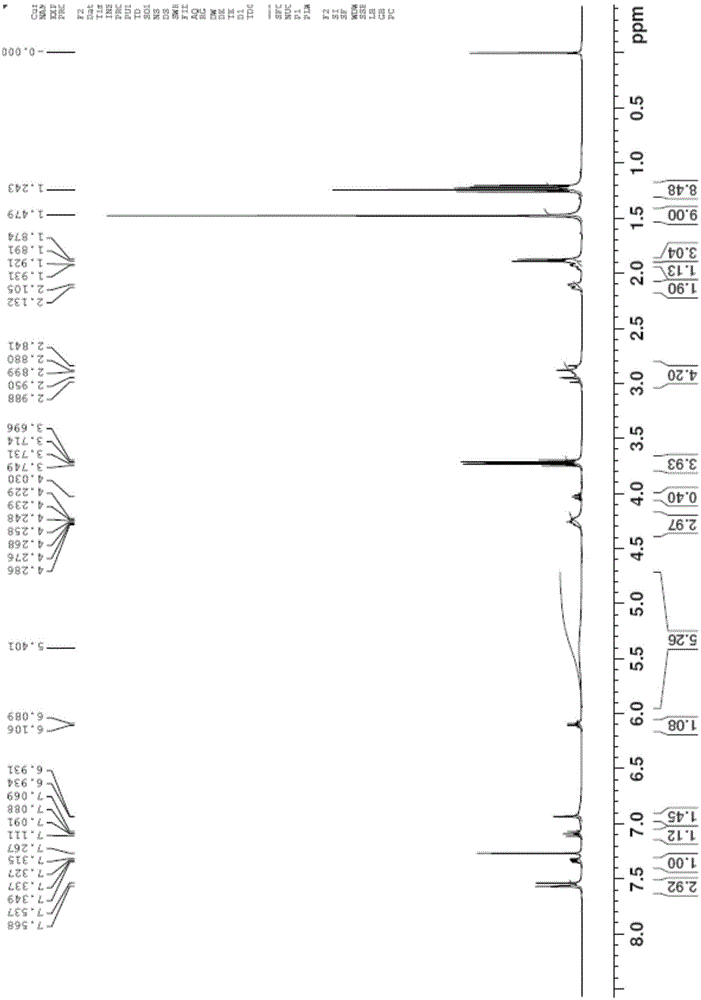
:克里唑替尼(Crizotinib)是美国辉瑞公司治疗肺癌的新药,这是第一个对间变性淋巴瘤激酶(ALK)进行靶向治疗的药品,可用于治疗ALK阳性的局部晚期或转移的非小细胞肺癌。其化学名称为:3-[(R)-1-(2,6-二氯-3-氟苯基)乙氧基]-5-[1-(哌啶-4-基)-1H-吡唑-4-基]吡啶-2-胺。具有如下式I结构的化合物为合成克里唑替尼的常用中间体,该化合物通常情况下在盐酸的作用下脱去Boc保护基即得到克里唑替尼,如可以参考美国专利申请US20060128724中提供的方法:目前式I化合物通常情况下由相应的硼酸酯与溴化合物发生Suzuki偶联反应制备得到,反应通常用到的催化剂为Pd(PPh3)2Cl2,如美国专利申请US20060128724中提供的方法,用方程式表示如下:或者如公开号为CN101735198的中国专利申请中公开的方法制备得到,无论是以哪种方式制备得到式I化合物,反应产物中都会残留有重金属Pd并且粗品有机杂质含量高,外观为黄色至棕黄色;通常情况下除去钯使用L-半胱氨酸盐酸盐、或者选择吸附剂,但对外观和有机杂质除去不明显。因此亟需开发一种新的式I化合物的纯化方法,以解决现有技术中存在的问题。技术实现要素:为了解决现有技术中利用常规Suzuki偶联方法制备得到式I化合物残留重金属钯含量高、外观差、有机杂质含量高等问题,本发明采用如下技术方案:一种具有下列式II结构的化合物:其中HA为有机酸,n为0.5或1。优选的HA为柠檬酸、草酸、富马酸、丁二酸、L-酒石酸、L-谷氨酸。更为优选的HA为柠檬酸或草酸。同时本发明提供了一种式II化合物的制备方法,为式I化合物与HA进行反应制备得到,其中HA和n的定义与上文相同。该反应可在合适的反应溶剂中进行,优选的反应溶剂为C1~C6醇类溶剂,苯类溶剂,醚类溶剂,腈类溶剂,酯类溶剂,C3~C6酮类溶剂。更为优选的反应溶剂为甲醇,乙醇,正丙醇,异丙醇,甲苯,甲基叔丁基醚,丙酮,四氢呋喃或二氧六环等。该反应适宜的反应温度为50-80℃。式II化合物在0-45℃析晶。其中所述的式I化合物与HA的摩尔用量比优选为1:(0.5~5),更有选为1:(0.5-1.1)。进一步地,利用本发明提供的式II化合物可以用于制备克里唑替尼。具体为式II化合物与盐酸反应,然后加入氢氧化钠制备得到克里唑替尼。优选的可以在反应过程中加入三苯基膦,能够进一步降低钯含量至小于10ppm。利用本发明提供的克里唑替尼中间体酸盐可以很好的除去反应底物式I化合物中的重金属钯残留,钯残留由1000ppm降至100ppm;并且除去部分有机杂质,利用本发明提供的式II化合物制备得到克里唑替尼,纯度高,重金属含量低。附图说明图1为按照实施例1的方法制备得到的3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶柠檬酸盐氢谱谱图。图2为按照实施例3的方法制备得到的3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧 基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶草酸盐氢谱谱图。图3为按照实施例4的方法制备得到的3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶富马酸盐氢谱谱图。图4为按照实施例7的方法制备得到的3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶L-谷氨酸盐氢谱谱图。图5为按照实施例1的方法制备得到的3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶柠檬酸盐HPLC谱图。图6为按照实施例6的方法制备得到的3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶对甲苯磺酸盐的HPLC谱图。具体实施方式为了更好的理解本发明的内容,下面结合具体实施例来做进一步的说明,但具体的实施方式并不是对本发明的内容所做的限制。本发明所使用的HPLC分离条件:仪器:Agilent1200色谱柱:ZORBAXEclipseXDB-C184.6mm×250mm×5μm柱温:30℃流动相A:称取1.36g磷的二氢钾,用去离子水溶解并稀释至1000mL,搅拌均匀;用85%磷酸调pH至2.8,混匀过滤,超声脱气。流动相B:乙腈梯度:时间(min)流动相A(%,V/V)流动相B(%,V/V)095530208035208035.195540955流速:1.0mL/min波长:UV220nm进样量:20μl对比实施例1:L-半胱氨酸盐酸盐/硅胶进行除钯实验取棕黄色3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺粗品(按照CN101735198提供的方法制备得到,HPLC纯度95.52%,钯残留1131ppm),加入L-半胱氨酸盐酸盐,搅拌5-7小时后,用氨水调pH至7,加入硅胶升温搅拌,处理析晶,得到棕色固体3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺L-半胱氨酸盐酸盐收率为82.85%,HPLC纯度95.54%,金属钯残留656.15ppm。实施例1:3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺柠檬酸盐的制备在常温下将异丙醇100ml和棕黄色3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺粗品(按照CN101735198提供的方法制备得到,HPLC纯度95.52%,钯残留1131ppm)(5.5g,0.01mol)投入反应瓶中,搅拌下投入柠檬酸一水物(2.1g,0.01mol);升温至65~75℃搅拌1小时;随后缓慢降温至40~45℃析晶,继续搅拌2~3小时;降温至5~20℃,保温搅拌2~4小时;保温结束,对其进行抽滤,抽滤结束,将湿品放入60~70℃真空干燥至少12小时,得灰白色固体3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶柠檬酸盐5.8g,收率89.78%,HPLC99.26%,钯残留153.117ppm,HPLC检测数据如下表1或见图5:表1编号保留时间宽度面积高度%面积121.04014.80025028.644444.470.1075222.39220.80052835.338357.740.2270322.71675.20023204116.261167292.2799.2556426.82019.60054536.147011.220.2343527.07220.00040872.694930.810.1756实施例2:3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺L-苹果酸盐的制备在常温下将异丙醇100ml和棕黄色3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺粗品(按照CN101735198提供的方法制备得到,HPLC纯度95.52%,钯残留1131ppm)(5.5g,0.01mol)投入反应瓶中,搅拌下投入L-苹果酸(1.34g,0.01mol);升温至60~75℃搅拌4小时;随后缓慢降温至40~45℃继续搅拌2~3小时;降温至 5~20℃,保温搅拌4-6小时;保温结束,对其进行抽滤,抽滤结束,将湿品放入60~70℃真空干燥至少12小时,得灰白色固体3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶L-苹果酸盐1.9g,收率29.77%,HPLC96.75%,钯残留244.235ppm。实施例3:3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺草酸盐的制备在常温下将异丙醇100ml和棕黄色3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺粗品(按照CN101735198提供的方法制备得到,HPLC纯度95.52%,钯残留1131ppm)(5.5g,0.01mol)投入反应瓶中,搅拌下投入草酸(0.97g,0.0077mol);升温至65~75℃搅拌2小时;随后缓慢降温至40~45℃析晶,继续搅拌2~3小时;降温至5~20℃,保温搅拌3~5小时;保温结束,对其进行抽滤,抽滤结束,将湿品放入60~70℃真空干燥至少12小时,得灰白色固体3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶草酸盐5.5g,收率89.70%,HPLC96.77%,钯残留197.994ppm。实施例4:3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺富马酸盐的制备在常温下将异丙醇100ml和棕黄色3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺粗品(按照CN101735198提供的方法制备得到,HPLC纯度95.52%,钯残留1131ppm)(5.5g,0.01mol)投入反应瓶中,搅拌下投入富马酸(0.64g,0.0055mol);升温至65~75℃搅拌4小时;随后缓慢降温至30~40℃析晶,继续搅拌2~3小时;降温至5~10℃,保温搅拌3~5小时;保温结束,对其进行抽滤,抽滤结束,将湿品放入60~70℃真空干燥至少12小时,得灰白色固体3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶富马酸盐5.45g,收率89.56%,HPLC97.72%,钯残留278.079ppm。实施例5:3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺L-酒石酸盐的制备在常温下将异丙醇100ml和棕黄色3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺粗品(按照CN101735198提供的方法制备得到,HPLC纯度95.52%,钯残留1131ppm)(5.5g,0.01mol)投入反应瓶中,搅拌下投入L-酒石酸(1.65g,0.011);升温至65~75℃搅拌3小时;随后缓慢降温至30~40℃析晶,继续搅拌5-6小时;降温至5~10℃, 保温搅拌4~5小时;保温结束,对其进行抽滤,抽滤结束,将湿品放入60~70℃真空干燥至少12小时,得灰白色固体3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶L-酒石酸盐4.65g,收率74.36%,HPLC95.99%,钯残留453.21ppm。实施例6:3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺对甲苯磺酸盐的制备在常温下将丙酮100ml和棕黄色3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺粗品(按照CN101735198提供的方法制备得到,HPLC纯度95.52%,钯残留1131ppm)(5.5g,0.01mol)投入反应瓶中,搅拌下投入对甲苯磺酸(1.89g,0.011);升温至60~70℃搅拌6小时;随后缓慢降温至30~40℃析晶,继续搅拌7-8小时;降温至5~10℃,保温搅拌4~5小时;保温结束,对其进行抽滤,抽滤结束,将湿品放入60~70℃真空干燥至少12小时,得灰白色固体3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶对甲苯磺酸盐5.01g,收率69.47%,HPLC64.8%,钯残留936.164ppm。HPLC检测数据如下表2或见图6:表2编号保留时间面积高度%面积19.067812888257038431.639214.065500335563821.947316.1171820626990.071420.138269513860.010522.36441355490.016623.71748185950.019724.1371664880474492564.800828.979384698439091.497实施例7:3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺L-谷氨酸盐的制备在常温下将异丙醇100ml和棕黄色3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺粗品(按照CN101735198提供的方法制备得到,HPLC纯度95.52%,钯残留1131ppm)(5.5g,0.01mol)投入反应瓶中,搅拌下投入L-谷氨酸(0.81g,0.0055);升温至65~75℃搅拌2小时;随后缓慢降温至30~40℃析晶,继续搅拌2-3小时;降温至5~10℃,保温搅拌2-3小时;保温结束,对其进行抽滤,抽滤结束,将湿品放入60~70℃ 真空干燥至少12小时,得灰白色固体3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶L-谷氨酸盐4.62g,收率74.1%,HPLC98.75%,钯残留304.866ppm。实施例8:3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺丁二酸盐的制备在常温下将异丙醇100ml和棕黄色3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺粗品(按照CN101735198提供的方法制备得到,HPLC纯度95.52%,钯残留1131ppm)(5.5g,0.01mol)投入反应瓶中,搅拌下投入丁二酸(1.3g,0.011);升温至65~75℃搅拌2小时;随后缓慢降温至30~40℃析晶,继续搅拌2-3小时;降温至5~10℃,保温搅拌2-3小时;保温结束,对其进行抽滤,抽滤结束,将湿品放入60~70℃真空干燥至少12小时,得灰白色固体3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶丁二酸盐4.62g,收率82.29%,HPLC99.38%,钯残留129.879ppm。实施例9-11是按照实施例1的方法同法操作,换用不同的反应溶剂及摩尔比(HA/反应底)所得到的试验结果:实施例14:克里唑替尼的制备在常温下,向反应瓶中加入二氯甲烷80mL,搅拌下依次投入3-[(1R)-1-(2,6-二氯-3-氟苯基)-乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶柠檬酸盐(3g,0.0046mol)、三苯基磷(1.31g,0.005mol)、纯化水50ml;滴加盐酸;滴毕,升温至35~45℃,保温反应1-2小时;反应完全,降温至20~25℃滴加30-60%氢氧化钠溶液5ml后搅拌4-5小时;反应完毕,抽滤,制得克里唑替尼湿品;将湿品进行干燥至少16小时后得到克里唑替尼1.91g,收率92.1%,HPLC99.83%,钯残留<10ppm。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1