衍生的超支化聚丙三醇的制作方法
本申请要求2010年3月1日提交的名称为“生物粘附性衍生的超支化聚丙三醇”的美国临时专利申请第61/309,304序列号的权益,通过引用将其全文并入。
技术领域
本发明涉及用于将药物或其他的生物活性部分递送至生物组织的治疗、它们的用途以及方法。具体地说,本发明涉及基于衍生的超支化聚丙三醇(dHPG)的聚合物以及用于治疗癌症、感染、和炎症性或自身免疫性疾病的方法。
背景
膀胱癌为第二最常见的泌尿生殖系恶性肿瘤。在最初诊断时,大约70%病例为非肌层侵润性的。目前用于浅表性疾病的治疗选择包括通过用导尿管将化学治疗剂膀胱内灌注至膀胱来局部地治疗膀胱癌。然而,这些治疗选择疗效有限。尽管接受了膀胱内化学疗法和/或免疫疗法,多达80%患有非肌层侵润性膀胱癌的患者发展为复发性肿瘤,其中20-30%发展成为更有侵略性的,潜在致死性肿瘤(Dalbagni,G.(2007)Nat.Clin.Pract.Urol.4:254-260)。
人们认为治疗失败部分地归因于有效对抗膀胱中膀胱癌细胞的药物的短驻留时间。例如,由于目前制剂在膀胱中不良的生物利用度,紫杉烷(taxanes)通常不用于膀胱内灌注。已有文献证明紫杉醇(Paclitaxel)在系统性膀胱癌治疗中有抗肿瘤活性,因为它以比诸如丝裂霉素C(mitomycin C)的水溶性药物的速率快20倍的速率渗透膀胱组织,甚至在灌注溶液除去后还允许治疗剂量延长的停留。然而,它的膀胱内用途受商品化制剂(TaxolTM)中CremophorTM-EL的存在所阻 碍,因为其使药物陷入水相环境并且减少了紫杉醇渗透至膀胱壁(Mugabe,C等.(2008)British J.Urology Int.102(7):978-986)。
当许多活性剂为疏水性或水不溶性时,它们经常需要基于水的或水相环境以用于众多适应症的有效治疗,所述适应症包括癌症(诸如肠癌、肺癌、膀胱癌以及泌尿生殖系统癌),感染(诸如消化道和气道的那些)以及炎症性或自身免疫性疾病(诸如刺激性膀胱、炎症性肠疾病以及慢性和急性炎症)。同样地,已研发了多系统用作此类制剂的递送媒介。这些系统的其中之一包括聚合物胶束的使用。
聚合物胶束为两性分子的,具有疏水性内核和亲水性外壳,正因为如此,它们可以由于疏水相互作用而将疏水性分子封入内核中。亲水性外壳保持系统可溶于水。然而,由于稀释效应或者环境因素,这些系统可能在膀胱中为不稳定的。
超支化聚丙三醇(“HPG”)为少数超支化聚合物的其中之一,所述超支化聚合物可以以可控的方式合成,具有预先确定的分子量和窄的多分散性(Kainthan,R.K.等.(2008)Biomacromolecules 9:886-895)。疏水性分子可被封入至HPG的疏水性内核(WO2006/130978)。
发明概述
本发明部分地基于以下发现:本文所述的衍生的超支化聚丙三醇(“dHPG”)可用作用于将药物或其他的生物活性部分递送至泌尿道(例如尿道和膀胱)、消化道(例如口、食道以及结肠)、气道(例如鼻和肺)、阴道腔和子宫颈以及腹膜腔的试剂,以治疗适应症,诸如癌症(例如,膀胱癌、胃癌、食道癌、肺癌、喉癌、口腔癌、窦癌、阴道癌或子宫颈癌)、感染(例如,消化道或气道感染),和炎症性或自身免疫性疾病(例如,刺激性膀胱、炎症性肠疾病或慢性或急性炎症)以及其他的适应症,其中要求将药物或其他的生物活性部分递送至组织或细胞。例如,本文所鉴定的聚合物可用于非肌层侵润性膀胱癌的灌注治疗。本文所鉴定的聚合物可显示粘膜粘附特性,该特性可用于非肌层侵润性膀胱癌的灌注治疗。
本文所述的dHPG可用作药物或其他的生物活性部分的载体并且 用于制备治疗药物,所述治疗药物用于将此类药物或部分递送至靶组织或细胞。具体地说,本文所述的dHPG可用作紫杉烷的载体,用于非肌层侵润性膀胱癌的治疗。
本文所述的凝聚的内核dHPG具有特别满足用于将药物递送至靶组织需要的令人惊奇的特性。具体地说,如本文所示,凝聚的内核dHPG的毒性更小并且具有更高的耐受性。
根据一实施方案,提供了超支化聚丙三醇,所述超支化聚丙三醇包含:内核,其包含用C1-C20烷基链衍生的超支化聚丙三醇,其中与内核的边缘相比,C1-C20烷基链与丙三醇单元的比率在内核中心更大;外壳,其包含至少一种与内核的羟基结合的亲水性取代基,其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。
根据另一实施方案,提供了将生物活性部分递送至生物组织的方法,所述方法包括:将负载生物活性部分的超支化聚丙三醇给予生物组织,其中超支化聚丙三醇包含:内核,其包含用C1-C20烷基链衍生的超支化聚丙三醇,其中与内核的边缘相比,C1-C20烷基链与丙三醇单元的比率在内核中心更大;外壳,其包含至少一种与内核的羟基结合的亲水性取代基,其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。该方法还包括将生物活性部分掺入超支化聚丙三醇。
根据其他的实施方案,提供了超支化聚丙三醇在用于将生物活性部分递送至生物组织中的用途,其中超支化聚丙三醇包含:内核,其包含用C1-C20烷基链衍生的超支化聚丙三醇,其中与内核的边缘相比,C1-C20烷基链与丙三醇单元的比率在内核中心更大;外壳,其包含至少一种与内核的羟基结合的亲水性取代基,其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。
根据另一实施方案,提供了超支化聚丙三醇在制备药物中的用途,所述药物用于将生物活性部分递送至生物组织,其中超支化聚丙三醇包含:内核,其包含用C1-C20烷基链衍生的超支化聚丙三醇,其中与内核的边缘相比,C1-C20烷基链与丙三醇单元的比率在内核中心更大; 外壳,其包含至少一种与内核的羟基结合的亲水性取代基,其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。
根据其他的实施方案,提供了包含超支化聚丙三醇与生物活性部分的药物组合物,其中超支化聚丙三醇包含:内核,其包含用C1-C20烷基链衍生的超支化聚丙三醇,其中与内核的边缘相比,C1-C20烷基链与丙三醇单元的比率在内核中心更大;外壳,其包含至少一种与内核的羟基结合的亲水性取代基,其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。
根据一实施方案,提供了超支化聚丙三醇,其包含内核,其包含用C1-C20烷基链衍生的超支化聚丙三醇;以及外壳,其包含至少一种与内核的羟基结合的亲水性取代基,其中至少一种亲水性取代基包含至少一种选自以下的官能团:-NH2,=NH2+,-NH3+,以及-NR3+,其中每一R为独立地C1-C6烷基或一个R为独立地C1-C6烷基并且两个R一起形成C3-C12环烷基,从而使得R3与氮形成季胺,而且其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。
根据其他的实施方案,提供了超支化聚丙三醇在用作预处理或联合治疗以增加组织中的药物摄取中的用途,超支化聚丙三醇包含:内核,其包含超支化聚丙三醇;以及外壳,其包含至少一种与内核的羟基结合的亲水性取代基,其中至少一种亲水性取代基包含至少一种选自以下的官能团:-NH2,=NH2+,-NH3+,以及-NR3+,其中每一R为独立地C1-C6烷基或一个R为独立地C1-C6烷基并且两个R一起形成C3-C12环烷基,从而使得R3与氮形成季胺,而且其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇,预处理。在一实施方案中,内核还可用C1-C20烷基链衍生。
根据另一实施方案,提供了用作预处理或联合治疗以增加组织中的药物摄取的超支化聚丙三醇,其包含:内核,其包含超支化聚丙三醇;以及外壳,其包含至少一种与内核的羟基结合的亲水性取代基,其中其中至少一种亲水性取代基包含至少一种选自以下的官能团:-NH2、=NH2+、-NH3+以及-NR3+,其中每一R为独立地C1-C6烷基或一个R为独立地C1-C6烷基并且两个R一起形成C3-C12环烷基,从而使得R3与氮形成季胺,而且其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇预处理。在一实施方案中,内核还可用C1-C20烷基链衍生。在一实施方案中,增加组织中的药物摄取可能引起组织的雨伞细胞的缺失。在一实施方案中,增加组织中的药物摄取可能没有引起组织的坏死和/或炎症。
根据另一实施方案,提供了超支化聚丙三醇,超支化聚丙三醇包含:内核,其包含来自于丙三醇环氧化物和C1-C20烷基环氧化物或C1-C20烷基缩水甘油醚聚合的超支化聚丙三醇,其中在所有或基本上所有的丙三醇环氧化物聚合之前,所有或基本上所有的C1-C20烷基环氧化物或C1-C20烷基缩水甘油醚为聚合的;以及外壳,其包含至少一种与内核的羟基共价结合的亲水性取代基,其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。根据其他的实施方案,提供了合成超支化聚丙三醇的方法,所述方法包括:聚合丙三醇环氧化物与C1-C20烷基环氧化物或C1-C20烷基缩水甘油醚,从而使得在所有或基本上所有的丙三醇环氧化物聚合之前,所有或基本上所有的C1-C20烷基环氧化物或C1-C20烷基缩水甘油醚为聚合的以形成超支化聚丙三醇;以及用至少一种亲水性取代基衍生超支化聚丙三醇的羟基,其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。丙三醇环氧化物可为缩水甘油。C1-C20烷基环氧化物可为1,2-环氧十八烷。C1-C20烷基缩水甘油醚可为C8-C10烷基缩水甘油醚。
根据另一实施方案,提供了超支化聚丙三醇,超支化聚丙三醇包含:内核,其包含用C1-C20烷基链衍生并负载多西紫杉醇(docetaxel)的超支化聚丙三醇;外壳,其包含至少一种与内核的羟基共价结合的亲水性取代基,其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。
根据其他的实施方案,提供了超支化聚丙三醇在用于将多西紫杉醇递送至生物组织中的用途,其中超支化聚丙三醇包含:内核,其包 含用C1-C20烷基链衍生并负载多西紫杉醇的超支化聚丙三醇;以及外壳,其包含至少一种与内核的羟基共价结合的亲水性取代基,其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。
超支化聚丙三醇可能还包括生物活性部分。超支化聚丙三醇可用作用于增加生物活性部分的药物摄取的预处理或联合治疗。生物活性部分可为一种或多种疏水性药物。生物活性部分可选自以下(或更多)其中一种:戊柔比星(valrubicin)、顺铂(cisplatin)、紫杉醇、多西紫杉醇。生物活性部分可为紫杉烷或其类似物。紫杉烷可为紫杉醇或其类似物。紫杉烷可为多西紫杉醇或其类似物。生物活性部分可为丝裂霉素或其类似物。丝裂霉素可包括所有的丝裂霉素类似物。丝裂霉素及其类似物可包括,例如,丝裂霉素A、丝裂霉素B、丝裂霉素C、丝裂霉素D、丝裂霉素F、丝裂霉素G、丝裂霉素H、丝裂霉素K以及其类似物。生物活性部分可为丝裂霉素C。生物活性部分可为丝裂霉素F。生物活性部分可为戊柔比星。生物活性部分可为长春花碱(vinblastine)。生物活性部分可为顺铂。生物活性部分可为甲氨蝶呤(methotrexate)。生物活性部分可为多柔比星(doxorubicin)或其类似物。生物活性部分可为表柔比星(epirubicin)。生物活性部分可为吉西他滨(gemcitabine)。生物活性部分可为依维莫司(everolimus)。生物活性部分可为苏拉明(suramin)。生物活性部分可为部分的组合。部分的组合可为甲氨蝶呤、长春花碱以及多柔比星(M-VAC)。部分的组合可为M-VAC与顺铂。
亲水性取代基可为聚乙二醇(PEG)(200-450g/ml),或甲氧基聚乙二醇(MPEG)(200-450g/ml),或其组合。至少一种亲水性取代基可为MePEG或PEG。至少一种亲水性取代基可为MePEG。至少一种亲水性取代基可为PEG。至少一种亲水性取代基可包含至少一种官能团,该官能团为-OH、-COOH、-NHS、-SH、-NH2、-NH3+或-NR3+,其中每一R为独立地C1-C6烷基或一个R为独立地C1-C6烷基并且两个R一起形成C3-C12环烷基,从而使得R3与氮形成季胺。
至少一种官能团可为-NH2、-NH3+或-NR3+,其中每一R为独立地C1-C6烷基或一个R为独立地C1-C6烷基并且两个R一起形成C3-C12环烷基,从而使得R3与氮形成季胺。至少一种官能团可为-NH2或-NH3+。至少一种官能团可为胺。可选择地,至少一种官能团可为-NH2。
超支化聚丙三醇可包含约1摩尔-约200摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约1摩尔-约100摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约1摩尔-约40摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约5摩尔-约40摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约10摩尔-约40摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约10摩尔-约30摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约30摩尔-约40摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约5摩尔-约15摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。所述至少一种亲水性取代基可结合约1%-约40%羟基。所述至少一种亲水性取代基可结合约5%-约30%羟基。所述至少一种亲水性取代基可结合约20%羟基。
可通过测量超支化聚丙三醇的分子量以及测量存在于超支化聚丙三醇总量中的亲水性取代基的量确定亲水性取代基的量/摩尔超支化聚丙三醇。本领域的技术人员应当理解,可利用不同的方法测量超支化聚丙三醇的分子量,例如,利用具有多角度激光散射探测的凝胶渗透色谱分析法。例如可通过滴定法测量存在于超支化聚丙三醇总量中的亲水性取代基的量。滴定法可为正向滴定法。滴定法可为反向滴定法。例如,其中亲水性取代基包含至少一种可为-NH2的官能团,抗诸如HCl酸的正向滴定法可用于测量亲水性取代基存在于HPG总量的量。其中亲水性取代基包含至少一种可为-NH2的官能团,可使用利用已知量的诸如HCl酸的反向滴定法并且滴定抗诸如NaOH的碱。例如可通过比色法测量存在于超支化聚丙三醇总量中的亲水性取代基的量。例如可通过荧光法测量存在于超支化聚丙三醇总量中的亲水性取代基的量。其中亲水性取代基包含至少一种可为-NH2的官能团,可使 用荧光胺测定。可通过多于一种方法测量存在于超支化聚丙三醇总量中的亲水性取代基的量以及通过多于一种方法所测量的亲水性取代基的量的平均值可用于计算亲水性取代基的摩尔/摩尔超支化聚丙三醇。其中亲水性取代基包含至少一种可为-NH2的官能团,荧光胺测定可为优选的方法以确定存在于超支化聚丙三醇总量中的亲水性取代基的量。
超支化聚丙三醇可包含约1摩尔-约200摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约1摩尔-约100摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约1摩尔-约40摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约5摩尔-约40摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约10摩尔-约40摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约10摩尔-约30摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约30摩尔-约40摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。超支化聚丙三醇可包含约5摩尔-约15摩尔的所述至少一种官能团/摩尔超支化聚丙三醇。
可通过测量超支化聚丙三醇的分子量以及测量存在于超支化聚丙三醇总量中的官能团的量确定官能团的量/摩尔超支化聚丙三醇。本领域的技术人员应当理解,可利用不同的方法测量超支化聚丙三醇的分子量,例如,凝胶渗透色谱分析法。例如,利用具有多角度激光散射探测的凝胶渗透色谱分析法测量超支化聚丙三醇的分子量。例如可通过滴定法测量存在于超支化聚丙三醇总量中的官能团的量。滴定法可为正向滴定法。滴定法可为反向滴定法。例如,其中所述至少一种官能团可为-NH2,抗诸如HCl酸的正向滴定法可用于测量-NH2存在于HPG总量的量。其中所述至少一种官能团可为-NH2,可使用利用已知量的诸如HCl酸的反向滴定法并且滴定抗诸如NaOH的碱。例如可通过比色法测量存在于超支化聚丙三醇总量中的官能团的量。例如可通过荧光法测量存在于超支化聚丙三醇总量中的官能团的量。其中所述至少一种官能团可为-NH2,可使用荧光胺测定。可通过多于一种方法 测量存在于超支化聚丙三醇总量中的官能团的量以及通过多于一种方法所测量的官能团的量的平均值可用于计算官能团的摩尔/摩尔超支化聚丙三醇。其中所述至少一种官能团可为-NH2,荧光胺测定可为优选的方法以确定存在于超支化聚丙三醇总量中的官能团的量。
C1-C20烷基链可为C5-C20烷基链。C1-C20烷基链可为C8-C18烷基链。C1-C20烷基链可为C8-C10烷基链。
至少一种亲水性取代基的一部分可位于内核中。
生物组织可为粘膜的膜。生物组织可为细胞。生物组织可为膀胱的尿路上皮表面。
如本文所述的dHPG可通过普通命名法来描述,所述普通命名法鉴定基础的超支化结构、内核属性、以及如下的表面属性:
HPG-内核(x)-外壳1(y1)-外壳2(y2)…-外壳n(yn) (I)
其指定了由超支化聚丙三醇组成的聚合物,其包含衍生的选自疏水基的取代基的内核,所述疏水基诸如C8、C10、C12或C18烷基,其为直链的或分支的或包含芳基取代基,其中内核取代基的量为x,用摩尔数或作为百分比表达。聚合物也具有外壳上n个取代基,诸如PEG或MePEG,或具有如本文所述的羧基(COOH)、羟基、胺(NR2)、N-羟基琥珀酰亚胺(NHS)、带电荷的胺(NR3+)、巯基(SH)等的取代基。每一外壳取代基可属于如存在于某一量y1、y2或yn并且用摩尔数或作为百分比表达。在一些符号中,通常的分类可属于相同的方式,而没有明确地鉴定每个的量。另外,当外壳取代基为PEG或MePEG时,还可通过这一聚合组分的链长度定义,例如MePEG350、PEG200等。然而对于通常的分类,分子量可以省略。
例如,HPG-C8/10-MePEG或HPG-C8/10-NH2分别属于内核(x)C8/10。术语“HPG-C8/10-MePEG”,诸如此类,在本文任何之处都可以与术语“HPG-C10-MePEG”互换地使用。在某些情况下,不鉴定内核(x)并且可以假设为C8/10。然而,可使用其他的具有C1-C20的烷基。根据其他的实施方案,提供了本文所述的dHPG在用于将药物递送至靶组织中的用途。根据其他的实施方案,提供了本文所述的dHPG在制备用于将药物递送至靶组织的药物中的用途。根据其他的实施方案,提供了本 文所述的dHPG用作预处理或联合治疗以增加组织中药物摄取中的预处理用途。根据其他的实施方案,提供了本文所述的dHPG用于治疗非肌层侵润性膀胱癌的用途。根据其他的实施方案,提供了本文所述的dHPG作为增加组织中用于治疗非肌层侵润性膀胱癌的药物摄取的预处理或联合治疗。根据其他的实施方案,提供了本文所述的dHPG在制备用于治疗非肌层侵润性膀胱癌的药物中的用途。非肌层侵润性膀胱癌的治疗可在哺乳动物中。哺乳动物可为人类。根据另一实施方案,提供了包含如本文所列的dHPG的药物组合物以及药学上可接受的赋形剂。根据其他的实施方案,提供了一种或多种本文所述的用于将药物递送至靶组织的dHPG。根据其他的实施方案,提供了用于制备本文所述的dHPG的方法。
本文所述的聚合物意指包括所有外消旋混合物和所有单独的结构同分异构体或变体,尤其是通过HPG结构内分支模式或在表面取代基与HPG物理连接方面所定义的。
附图简要说明
图1显示了多西紫杉醇(DTX)样品的超高效液相色谱(UPLC)的色谱图。
图2显示了DTX洗脱液和它的差向异构体(7-表-DTX)的峰面积与时间的函数以确定如本文所述的dHPG掺入DTX的稳定性。
图3A显示了DTX与7-表-DTX离子(+H+)m/z 808.5在掺入DTX的HPG-C8/10中单一的离子记录(SIR)。
图3B显示了DTX与7-表-DTX离子(+H+)m/z 808.5在掺入DTX的HPG-C8/10-MePEG-NH2中的SIR。
图4显示了对掺入DTX的HPG-C8/10-MePEG-NH2而言的总离子流(TIC)。
图5显示了针对DTX所提出的现有技术破碎模式。
图6显示了对pH7.4和pH6.0时掺入DTX的HPG-C8/10-MePEG-NH2而言,DTX(和7-表-DTX)洗脱液的峰面积与时间的函数。
图7A显示了对正常的内核(NC)制剂和凝聚的内核(CC)制剂而言,KU7细胞增殖的百分比与HPG-C8/10的浓度的函数。
图7B显示了对于正常的内核(NC)制剂和凝聚的内核(CC)制剂而言,KU7细胞增殖的百分比与HPG-C8/10-MePEG的浓度的函数。
图8显示了KU7细胞增殖的百分比与聚合物浓度的函数以确定各种dHPG的生物相容性。
图9显示了HPG-C8/10在分别代表树状的、直链的1-3、以及直链的1-4单元的D6-DMSO.D、L13以及L14中的异核单量子相关谱(HSQC)光谱和400MHz质子(顶部)。
图10显示了(A)HPG-C8/10-MePEG6.5的400MHz HSQC质子(顶部)和HSQC光谱以及(B)MePEG350环氧化物、O/DGE、以及HPG-C8/10-MePEG13聚合物的叠加的400MHz HSQC光谱。
图11显示了(A)PTX酯键的碱催化的乙醇分解以产生巴卡亭(Baccatin)III以及它的侧链乙酯(N-苯甲酰-3-苯基异丝氨酸乙酯)和(B)示例在利用未纯化的HPG-C8/10-MePEG制备制剂中通过UPLC-MS/MS测定PTX降解产物的鉴定的代表性色谱图。
图12显示了示例HPG-C8/10-MePEG13的纯化对通过UPLC UV分析所测量的PTX的化学稳定性(2.1分钟的停留时间)的影响的代表性色谱图。(A)PTX的色谱图,该PTX配制于磷酸盐缓冲液(PBS)(pH7.4)中新鲜组成的未纯化的HPG中,(B)(A)中相同的制剂,老化48h的色谱图,以及(C)PTX的色谱图,该PTX配制于PBS(pH7.4)中新鲜组成的纯化的HPG中。
图13显示了人造尿液中37℃下自HPG-C8/10-MePEG释放的PTX与DTX。(A)自HPG-C8/10-MePEG(6与13mol)释放的累积的DTX。(B)自人造尿液中HPG-C8/10-MePEG(pH4.6与6.5)释放的累积的PTX。
图14显示了示例1h暴露后HPG-C8/10-MePEG13-TMRCA纳米颗粒完全摄取的KU7细胞的共聚焦荧光成像。(A)具有苯基吲哚(DAPI)染色未处理的KU7细胞允许细胞核可视为蓝色(如图中白色所示)。(B)已与HPG-C8/10-MePEG13-TMRCA纳米颗粒孵育1h的KU7细胞。
图15显示了商品化制剂与抗KU7-荧光素酶细 胞系的负载PTX和/或DTX的HPG-C8/10-MePEG纳米颗粒、低级的(RT4,MGHU3)与高级的(UMUC3)人尿路上皮癌细胞系的体外细胞毒性效应。
图16显示了膀胱内紫杉烷制剂对原位膀胱癌异种移植的治疗效应。媒介对照(PBS&空白的HPG-C8/10-MePEG)、(1mg/ml,Bristol-Myers-Squibb)、(0.5mg/ml,Sanofi-Aventis)或负载紫杉醇(PTX,1mg/ml)或多西紫杉醇(DTX,0.5mg/ml)的HPG-C8/10-MePEG。
图17显示了来自不同的治疗组,取自随机某天、18天以及33天的小鼠的生物荧光成像的代表性序列。右侧,相同的小鼠在PBS对照、 以及负载DTX的HPG-C8/10-MePEG治疗组中的代表性膀胱横截面。
图18显示了最终收获的膀胱的代表性组织切片,其来自接受了各种处理的小鼠:A)PBS,B)(1mg/ml),C)HPG-C8/10-MePEG/PTX(1mg/ml),D)(0.5mg/ml),E)HPG-C8/10-MePEG/DTX(0.5mg/ml),F)HPG-C8/10-MePEG(无药物)。
图19显示了(A)HPG-C8/10-MePEG的一维质子谱(顶部曲线)和HSQC光谱,和(B)在9.4T磁场强度处获得的HPG-C8/10-MePEG-NH2(121)的一维质子谱(顶部曲线)和HSQC光谱。
图20显示了通过粘蛋白-颗粒方法所评估的HPG的粘膜粘附特性。
图21显示了(A)若丹明(rhodamine)标记的HPG的体外Ku7-荧光素酶结合以及(B)暴露于HPG溶液的Ku7-荧光素酶细胞的细胞活力。
图22显示了自HPG-C8/10-MePEG与pH6.5人造尿液中HPG-C8/10-MePEG-NH2纳米颗粒释放的累积的DTX。
图23显示了(A)来自除了PBS对照以外每一治疗组取自肿瘤接种后2天、8天、12天的小鼠的生物荧光成像以及(B)单一的膀胱内DTX制剂对原位膀胱癌异种移植的处理效应。右侧图板,除了PBS和 (0.5mg/ml)组以外治疗组的详细视图。
图24显示了下列负载0.2mg/ml DTX的HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2(121)单一膀胱内治疗的小鼠的生物荧光成像。
图25显示了抗KU7荧光素酶细胞的DTX制剂以及低级的(RT4,MGHU3)与高级的(UMUC3)人尿路上皮癌细胞系二者的体外细胞毒性。
图26显示了单一的膀胱内DTX制剂对原位膀胱癌异种移植的治疗效应。小鼠的生物荧光成像显示于左侧面板。
图27显示了研究结束时,来自于接受了0.2mg/ml DTX的各种制剂:(A)HPG-C8/10-MePEG-NH2,(B)HPG-C8/10-MePEG,(C)的小鼠所收获的膀胱的代表性组织切片。治疗组中,A、B和C,数字1-3属于不同的放大率。
图28显示了与DTX(Taxotere TM)和PTX(TaxolTM)的商品制剂相比,对掺入DTX或紫杉醇(PTX)的HPG-C8/10-MePEG而言,肿瘤生物荧光与时间的函数。
图29显示了HPG-C8/10-MePEG或HPG-C8/10-MePEG-NH2中50μg DTX灌注后2小时DTX在膀胱内的停留。
图30显示了用PBS、游离的若丹明(TMRCA)、HPG-C8/10-MePEG标记的若丹明(HPG-C8/10-MePEG-TMRCA)、HPG-C8/10-MePEG-NH2标记的若丹明(HPG-C8/10-MePEG-NH2-TMRCA)灌注的原位膀胱癌。(B)膀胱肿瘤内的荧光量,(C)肿瘤组织中所观察到的若丹明荧光与距离膀胱腔的函数。
图31显示了氘代甲醇(methanol-d4)中HPG-C8/10-MePEG-COOH的(A)1H核磁共振谱与(B)13C核磁共振谱。
图32显示了HPG聚合物中的结构单元。每一树状的(D)、末端的(T)以及直链(L13或L14)单元以主要的(p)和次要的(s)单元存在。对未修饰的聚合物而言,R)HPG,对未修饰的聚合物而言,R)HPG、C8/10、MePEG或COOH。编号方案适用于Dp单元。
图33显示了(A)HPG-C8/10-OH、(B)HPG-C8/10-COOH(高的COOH)、以及(C)HPGC8/10-MePEG6.5的代表性多重编辑的HSQC光谱。光谱表明了代表性分配。
图34显示了(A)HPG-C8/10-OH与(B)HPG-C8/10-COOH的HSQC光谱的扩展区域。给出了代表性分配。
图35显示了HPG-C8/10-MePEG6.5(顶部)、HPG-C8/10-MePEG6.5-COOH113(中心)以及HPG-C8/10-MePEG6.5-COOH348(底部)的FT-IR光谱。
图36显示了结合顺铂的HPG-C8/10-MePEG-COOH的代表性结构。
图37显示了顺铂与(空白的三角)HPG-C8/10-MePEG6.5-COOH113或(实心的方块)HPG-C8/10-MePEG6.5-COOH348在调至pH6.0的蒸馏水中的结合。
图38显示了在药物浓度1mg/ml和聚合物浓度10mg/ml时游离的顺铂(空白的菱形)或与(A)HPG-C8/10-MePEG6.5-COOH113或(B)HPG-C8/10-MePEG6.5-COOH348结合的顺铂的体外释放。释放介质为37℃下pH4.5(实心的方块)、6.0(空白的三角)、7.4(倒三角)的1mM PBS或人造尿液(空白的方块)。
图39显示了与HPG-C8/10-OH(实心的方块)、HPG-C8/10-MePEG6.5(空白的三角)、HPGC8/10-MePEG6.5-COOH113(实心的圆圈)以及HPG-C8/10-MePEG6.5-COOH348(空白的菱形)孵育(A)2小时和(B)72小时后KU-7-荧光素酶细胞的细胞活力。
图40显示了与游离的顺铂(实心的圆圈)、负载顺铂的HPG-C8/10-MePEG6.5-COOH113(空白的三角)以及HPG-C8/10-MePEG6.5-COOH348(实心的方块)孵育(A)2小时和(B)72小时后KU-7-荧光素酶细胞的活力。
图41显示了暴露于含0.5mg/ml DTX(实心的圆圈)的Tween 80、含0.5mg/ml DTX的HPG-C8/10-MePEG-NH2(37摩尔胺/聚合物摩尔)(空白的方块)、含0.5mg/ml DTX的HPG-C8/10-MePEG-NH2(10摩尔胺/聚合物摩尔)(空白的三角)、含0.5mg/ml的DTXHPG-C8/10-MePEG(实心的倒三角)后猪膀胱组织内DTX的组织横向-纵向曲线。四种制剂的重复游程数的平均值,与Tween 80相比(例如Taxotere制剂),利用具有递增的胺含量的HPG聚合物比较渗透力。每一游程数中,进行5-6次重复次数。
图42显示了伴随及无预处理1小时暴露于不同的DTX制剂后猪膀胱组织内DTX的组织横向-纵向曲线。含0.5mg/ml DTX的Tween 80 伴随壳聚糖预处理(空白的圆圈),含0.5mg/ml DTX的Tween 80伴随HPG-C8/10-MePEG-NH2预处理(空白的菱形),含0.5mg/ml DTX的HPG-C8/10-MePEG-NH2无预处理(空白的倒三角),以及含0.5mg/ml DTX的HPG-C8/10-MePEG伴随壳聚糖预处理。
图43显示了对伴随及无预处理1小时不同的DTX制剂而言DTX的AUCs。含0.5mg/ml DTX的Tween 80伴随壳聚糖预处理,含0.5mg/ml DTX的Tween 80伴随HPG-C8/10-MePEG-NH2预处理,含0.5mg/ml DTX的HPG-C8/10-MePEG-NH2无预处理,含0.5mg/ml DTX的HPG-C8/10-MePEG-NH2伴随壳聚糖预处理,以及含0.5mg/ml DTX的HPG-C8/10-MePEG伴随壳聚糖预处理。在post-hoc Tukey分析中,有意义的单因素方差分析结果p=0.0007后,线条(bars)表示组间无显著性差异(p>0.05)。误差线表示S.E.M。
图44显示了伴随预处理1小时暴露于丝裂霉素制剂后丝裂霉素F在猪膀胱组织中的组织横向-纵向曲线。伴随HPG-C8/10-MePEG-NH2(10摩尔胺/HPG摩尔)预处理的丝裂霉素F(空白的方块)以及伴随HPG-C8/10-MePEG-NH2(37摩尔胺/HPG摩尔)预处理的丝裂霉素F(实心的菱形)。
图45显示了用HPG递送媒介:对照(台氏(Tyrode’s)缓冲液,壳聚糖&具有0摩尔胺/摩尔聚合物的HPG-C8/10-MePEG)体外处理的猪膀胱的SEM图像。
图46显示了用HPG递送媒介:HPG-C8/10-MePEG-NH2 10&37摩尔胺/摩尔聚合物,0.1,1&10%w/v溶液体外处理的猪膀胱的SEM图像。
图47显示了用PBS灌注2小时处理的小鼠膀胱的表面的SEM图像。图像取自2小时灌注时间段后就收获的膀胱。
图48显示了用HPG-MePEG 10%溶液灌注2小时处理的小鼠的膀胱的表面的SEM图像。图像取自2小时灌注时间段后,灌注后B)6以及C)24小时后就收获的膀胱。
图49显示了用HPG-MePEG-NH2(10mol/mol)10%溶液灌注2小时处理的小鼠的膀胱的表面的SEM图像。图像取自A)2小时灌注时间段 后,灌注后B)6以及C)24小时后就收获的膀胱。箭头表示单一的雨伞细胞缺失,暴露了上皮的底层。
图50显示了用HPG-MePEG-NH2(37mol/mol)1%溶液灌注2小时处理的小鼠的膀胱的表面的SEM图像。图像取自A)2小时灌注时间段后,灌注后B)6以及C)24小时后就收获的膀胱。
图51显示了用HPG-MePEG-NH2(37mol/mol)10%溶液灌注2小时处理的小鼠的膀胱的表面的SEM图像。图像取自A)2小时灌注时间段后,灌注后B)6以及C)24小时后就收获的膀胱。
图52显示了在除去灌注导管的点上(2小时,对所有的组而言N=6)以及在膀胱收获时(2、6、24小时,对6与24小时取样时间而言n=1-3)自小鼠所收获的尿液中的细胞计数。
图53显示了在A)HPG-MePEG 10%溶液,B)HPG-MePEG-NH2(低的)10%溶液,C)HPG-MePEG-NH2(高的)1%溶液,D)HPG-MePEG-NH2(高的)10%溶液,CN)PBS缓冲液(对照)灌注后2、6和24小时小鼠血液中以及U)未处理的动物循环的TNFα水平。结果以用于构建标准曲线的标准液显示。虚线代表最低标准液的信号(0.6pg/mL标准液浓度)。
发明的详细描述
本文所述的新的聚合物包括通式I中所示的那些,其所有似乎都与HPG有关系。早先已描述了HPG的合成,其包括两性共聚物与两性嵌段共聚物的产生,所述共聚物的产生包括与各种官能团的衍生物和/或共聚物与嵌段共聚物的产生(诸如烷基通过酯键的添加和聚亚烷基乙二醇基的添加)。描述HPG制备的出版物包括:美国专利5,112,876;美国专利6,469,218;美国专利6,765,082;美国专利6,822,068;WO 2000/77070;Sunder,A.等,(1999)Macromolecules 32:4240-46,(2000)Macromolecules 33:309-14,(2000)Macromolecules 33:1330-37,以及(2000)Adv.Mater 12:235-239;Knischaka,R.等,(2000)Macromolecules 33:315-20;Haag,R.,等,(2000)Macromolecules 33:8158-66,以及(2002)J.Comb.Chem.4:112-19;Kautz,H.等,(2001) Macromol.Symp.163:67-73;Karger-Kocsis,J.等,(2004)Polymer,45:1185-95;Gao,C.&Yan,D.(2004)Prog.Polym.Sci.29:183-275;以及Tziveleka,L等(2006)Macromol.Biosci.6:161-169)。Sunder,A等,(1999)Angew.Chem.Int.Ed.38:3552-55包含两性修饰的HPG的制备以及此类聚合物的衍生的描述,所述衍生包括与各种取代基及官能团的衍生。
本文所述的dHPG可包括C1-C30烷基链或其他类似的烷基链。然而,本文所述的dHPG也可包括C1-C20烷基链或其他类似的烷基链。术语“烷基”如本领域技术人员通常所理解的使用并且经常指的是具有1个-20个碳原子的单价饱和的脂肪族的烃基,除非另有定义。碳氢化合物可为直链的或支链的并且可包含脂环族的或芳基取代基。烷基链可选自C1-C20烷基链的其中之一或更多。可选择地,烷基链可选自C2-C19或C3-C18或C4-C17烷基链的其中之一或更多。可选择地,烷基链可选自C5-C16或C6-C15或C7-C14烷基链的其中之一或更多。可选择地,烷基链可选自C8-C13或C9-C12或C10-C15烷基链的其中之一或更多。可选择地,烷基链可选自C5-C15或C5-C10或C5-C20烷基链的其中之一或更多。烷基链或对内核而言所选的链可取决于对dHPG的预期用途。例如,对负载紫杉醇而言,C18烷基以及C8/10不起效。
本文所述的HPG可包括具有官能团的取代基衍生的HPG从而使得衍生的HPG(“dHPG”)为粘膜粘附的,一般而言,为生物粘附的。在该术语最通常的意义中,dHPG将形成键或与生物组织相互作用,所述生物组织可为细胞或细胞外物质。键或相互作用可为任何类型的,其包括范德华(van der Waals)相互作用、氢键、静电相互作用、离子键或共价键。
术语“粘膜粘附”或“粘膜粘附的”如本领域技术人员通常所理解的使用并且经常指的是发生在聚合物材料与生物组织间的粘附现象,所述生物组织可以包括细胞表面、细胞表面的粘液或覆盖粘膜的粘液凝胶层。因为粘蛋白存在于膀胱的尿路上皮表面,所以含有粘膜粘附官能团的dHPG可用于将靶向药物递送至膀胱表面以及其他的粘膜。
通常,本文所述的dHPG具有其包括的“内核”,启动因子(例如三 羟甲基丙烷(TMP))以及超支化聚丙三醇。在一实施方案中,超支化聚丙三醇内核可用C1-C20烷基链衍生。在一实施方案中,“内核”可裹入“外壳”中,其中外壳包含至少一种与内核的羟基结合的亲水性取代基,并且其中超支化聚丙三醇包含约1摩尔-约200摩尔的所述至少一种亲水性取代基/摩尔超支化聚丙三醇。
本文所用的“启动因子”定义为包含烷基组分以及多于一个,但是优选多于两个羟基的小分子。然而,启动因子可具有三个或四个或更多的羟基。启动因子的一个实例为三羟甲基丙烷(TMP)。
本文所用的“凝聚的内核”定义为其中与内核的边缘相比,C1-C20烷基链与丙三醇单元的比率在内核中心更大的内核。例如,C10烷基链可掺入至该结构中从而使得遍及整个的超支化结构,相对于丙三醇,它并不是均匀分布的,而是它的分布使得它在超支化内核结构的中心(例如邻近启动因子)中比靠近它的边缘紧邻于外壳的取代基更凝聚。内核体系结构为“凝聚的”的程度可通过试剂的添加而控制。术语“中心的”可定义为具有零体积点的精确的中心。可选择地,dHPG可具有半径“r”,其中具有半径“rc”的中心体积中烷基与丙三醇的比率比dHPG整体中烷基与丙三醇的总比率更大,其中rc<r。
对“常规的”或“正常的”内核而言,丙三醇环氧化物(超支化组分单体)与烷基环氧化物(其给予内核疏水性)于反应自始至终可以恒定的比率增加。对凝聚的内核而言,烷基环氧化物在反应的最早期以较高的比例增加并且在反应的后期以较低的比例(低至零)减少。这一减少可连续地发生或以不连续的阶段发生,存在着两个不连续的阶段的最小量。
内核体系结构可在组分添加的比率方面定义。例如,凝聚的内核聚合物可以多步骤合成,每一步骤具有确定比率的内核单体,一个为丙三醇环氧化物并且另一个为疏水性烷基环氧化物。可通过早期步骤中添加与后期或最后步骤中所添加的组分的比率相比具有更高比率的烷基环氧化物制造凝聚的内核分子。可选择地,比率可能在随时间进程中改变,从而使得对反应期间第一的(或早期的)时间段而言,添加更高的烷基环氧化物与丙三醇环氧化物的比率,所述比率比后期时间 段中所添加的比率高。在这一方法中,比率在整个反应中随着梯度不断地变化。
聚合物剩余的羟基可用诸如MePEG或PEG的其他亲水性取代基“衍生”以形成亲水性外壳,其包括具有羟基、羧基、胺(其包括一级的、二级的以及三级的胺)NHS、醚、巯基、卤素、硫醚、酯、硫酯、酰胺、琥珀酰亚胺以及其他类似的官能团的取代基。外壳形成期间,外壳取代基的部分可能与位于朝着聚合物中心的羟基反应。即使此类反应发生,内核仍维持它的疏水性特征。
如本文所用的,术语“两性的”或“两性的聚合物”如本领域技术人员通常所理解的使用并且经常指的是单一的分子中疏水性与亲水性部分二者的存在。疏水性指的是任一物质或其部分,其在非极性溶剂中比在极性溶剂中更可溶的。可通过分子中包含非极性基团给予疏水性,所述非极性基团包括但不限于长链饱和的与不饱和的脂肪族烃基团并以及通过一个或多个芳族的、脂环族的或杂环的基团所取代的此类基团。亲水性指的是任一物质或其部分,其在极性溶剂中比在非极性溶剂中更可溶的。亲水性特征衍生自极性或带电荷的基团的存在,所述极性或带电荷的基团诸如碳水化合物、磷酸酯、羧基的、硫酸根、氨基的、巯基、硝基、羟基以及其他类似的基团。亲水性部分可包含MePEG、胺、羧酸或NHS。
术语“聚乙二醇”或“PEG”如本领域技术人员通常所理解的使用并且经常指的是具有分子量约200-约20,000的此类化合物,所述分子量取决于聚合物链中环氧乙烷单元的数量。优选的分子量为约200-约400,约200-约1000以及约200-约2000,尽管约2000-约8000的分子量也可使用。
术语“甲氧基聚(环氧乙烷)”或“MePEG”如本领域技术人员通常所理解的使用并且经常指的是具有约350-约10,000分子量的此类化合物,所述分子量取决于聚合物链中环氧乙烷的数量。优选的分子量为约350-约550,约350-约750,以及约350-约2000,尽管约2000-约5000的分子量也可使用。
短语“局部的或靶向递送”如本领域技术人员通常所理解的使用并 且经常指的是化合物直接递送至有机体中靶位点。
在一些实施方案中,如本文所述的dHPG可用于局部的或靶向治疗泌尿道(例如尿道和膀胱)、消化道(例如口、食道以及结肠)、气道(例如鼻和肺)、阴道腔和子宫颈以及腹膜腔的适应症以治疗适应症,诸如癌症(例如,膀胱癌、胃癌、食道癌、肺癌、喉癌、口腔癌、窦癌、阴道癌或子宫颈癌)、感染(例如,消化道或气道感染),和炎症性或自身免疫性疾病(例如,刺激性膀胱、炎症性肠疾病或慢性或急性炎症)以及其他的适应症,其中要求将药物或其他的生物活性部分递送至组织或细胞。例如,如本文所述的dHPG可用于非肌层侵润性膀胱癌的局部的或靶向治疗。在一些实施方案中,如本文所述的聚合物可用于药物或组合物的制备,所述药物或组合物用于一种或多种本文所列的适应症(例如非肌层侵润性膀胱癌)的局部或靶向治疗。本发明的一些方面使用包含本文所述的dHPG的组合物与药学上可接受的赋形剂或载体。也提供了治疗一种或多种本文所列的适应症(例如非肌层侵润性膀胱癌)的方法。此类方法可包括给予需要其个体如本文所述的dHPG或如本文所述的dHPG的组合物,或如本文所述的dHPG或如本文所述的dHPG的组合物的有效量,其中将生物活性剂掺入dHPG。
在一些实施方案中,如本文所述的dHPG可用作预处理或联合治疗以增加组织内药物摄取。在一些实施方案中,如本文所述的dHPG可用作预处理或联合治疗以增加药物的药物摄取,所述药物用于局部的或靶向治疗泌尿道(例如尿道和膀胱)、消化道(例如口、食道以及结肠)、气道(例如鼻和肺)、阴道腔和子宫颈以及腹膜腔的适应症以治疗适应症,诸如癌症(例如,膀胱癌、胃癌、食道癌、肺癌、喉癌、口腔癌、窦癌、阴道癌或子宫颈癌)、感染(例如,消化道或气道感染),和炎症性或自身免疫性疾病(例如,刺激性膀胱、炎症性肠疾病或慢性或急性炎症)以及其他的适应症,其中要求将药物或其他的生物活性部分递送至组织或细胞。例如,如本文所述的dHPG可作为预处理或联合治疗使用以增加用于局部或靶向治疗非肌层侵润性膀胱癌的药物的药物摄取。在一些实施方案中,如本文所述的dHPG可作为用于增加组织内药物摄取的预处理使用。在一些实施方案中,如本文所述的dHPG 可作为用于增加组织内药物摄取的联合治疗使用。在一实施方案中,作为联合使用的dHPG的用途可包括其中在用药物或生物活性部分治疗期间,药物或生物活性部分没有负载在dHPG中。在一实施方案中,作为联合使用的dHPG的用途可包括其中在用药物或生物活性部分治疗期间,部分或所有的药物或生物活性部分都负载在dHPG中。在一些实施方案中,如本文所述的dHPG可作为用于增加组织内药物摄取的预处理和联合治疗使用。在一些实施方案中,如本文所述的dHPG可作为用于增加组织内药物摄取的预处理和联合治疗使用,与缺少预处理或联合治疗时组织中药物摄取比较。在一些实施方案中,如本文所述的dHPG可作为用于增加组织内药物摄取的预处理和联合治疗使用而没有引起组织的坏死和/或炎症。在一些实施方案中,增加组织内药物摄取可包括引起雨伞细胞缺失。在一些实施方案中,增加组织内药物摄取可包括引起雨伞细胞缺失而没有引起组织的坏死和/或炎症。在一些实施方案中,雨伞细胞可为膀胱的尿路上皮表面的雨伞细胞。“增加药物摄取”的表达如本领域技术人员通常所理解的使用并且经常指的是增加细胞或组织中药物的浓度或积聚。
在一些实施方案中,可在药物摄取的平均浓度(Cavg)增加方面测量药物摄取增加,利用作为预处理或联合治疗的dHPG,与没有预处理或联合治疗时药物摄取的浓度比较。本领域技术人员应理解可在组织深度的不同范围或点上测量浓度。例如可在组织深度的0>3350,0>1500,200>3350,或200>1500μm范围测量浓度。在一实施方案中,伴随作为预处理或联合治疗的dHPG使用,药物摄取的浓度可增加1.3-4.0,1.8-2.8,1.3-2.4,2.0-2.6,1.5,1.8,2,2.2,2.4,2.6,2.8,3.0,3.2,3.4,3.6,3.8或4.0倍,与没有预处理或联合治疗时药物摄取的浓度比较。在一些实施方案中,可在药物摄取的峰值浓度(Cmax)增加方面测量药物摄取增加,利用作为预处理或联合治疗的dHPG,与没有预处理或联合治疗时药物摄取的浓度比较。在一实施方案中,伴随作为预处理或联合治疗的dHPG使用,药物摄取的浓度可增加1.3-4.0,1.8-2.8,1.3-2.4,2.0-2.6,1.5,1.8,2,2.2,2.4,2.6,2.8,3.0,3.2,3.4,3.6,3.8或4.0倍,与没有预处理或联合治疗时药物摄取的峰值浓度比较。在一些实 施方案中,可在药物摄取的曲线下面积(AUC)(x-y)增加方面测量药物摄取增加,利用作为预处理或联合治疗的dHPG,与没有预处理或联合治疗时药物摄取的曲线下面积(x-y)比较。本领域技术人员应理解可在组织深度的不同范围或点上测量曲线下面积(x-y)。例如可在组织深度的0>无限,0>3350,0>1500,200>无限,200>3350或200>1500μm范围测量曲线下面积(x-y)。在一实施方案中,伴随作为预处理或联合治疗的dHPG使用,药物摄取的曲线下面积(x-y)可增加1.3-4.0,1.8-2.8,1.3-2.4,2.0-2.6,1.5,1.8,2,2.2,2.4,2.6,2.8,3.0,3.2,3.4,3.6,3.8或4.0倍,与没有预处理或联合治疗时药物摄取的曲线下面积(x-y)比较。本领域技术人员应理解存在用于测量药物摄取增加的可选择的方法,例如渗透性增强比,R,计算方法为预处理或联合治疗存在以及不存在时渗透性的系数,如可使用Grabnar等,(International Journal of Pharmaceutics 256(2003)167-173)所报道的。
在一些实施方案中,如本文所述的dHPG可以为溶剂添加形式。dHPG可与溶剂、水和/或缓冲液的非化学计量有关,通常表示成重量或容积百分比。例如,并且不限于,溶剂可为药学上可接受的溶剂或其他的生物相容性溶剂,其包括乙醇、二甲基亚砜(DMSO)、丙二醇、丙三醇、聚乙二醇200(PEG200)、聚乙二醇300(PEG300)、环氧二元醇(Transcutol)或Solutol。
如本文所述的dHPG包括所有可能的立体化学的替代选择,其包括示例或本文所述的那些。
在一些实施方案中,如本文所述的dHPG包括诸如具有不同支链模式的几何异构体的同分异构体。通过本文所公开的方法合成的dHPG为随机分支的HPG并且将包含丙三醇单体,所述丙三醇单体为充分反应的,例如以三个方向连接,或部分反应的,以一个或两个方向连接另外的单体。可通过分析技术(例如2D NMR HSQC实验)确认每一分支的体系结构的存在。
根据本文所述的一些实施方案,组合物与dHPG可以任一各种已知的途径给药。dHPG可作为下列形式给药:膀胱内计量溶液或其他的引起灌洗功能的组合物(其包括口服灌洗、腹膜内冲洗溶液或用于鼻 或阴道腔的冲洗)、滴眼剂、待被吞食的口服溶液、气雾剂、或作为喷雾用于吸入的溶液、或作为半固体待被插入至紧邻诸如粘膜表面的生物组织。
应理解限制本文所述的掺入药物或其他的生物活性剂的dHPG递送至靶组织或要求药物递送的细胞是潜在有利的。例如预期所述的掺入生物活性剂的dHPG选择性递送至患有或怀疑患有非肌层侵润性膀胱癌个体的膀胱尿路上皮表面可提供治疗效应而不在身体的其他组织中产生显著的副作用。可适用于如本文所述的掺入紫杉烷的dHPG给药的一方法实例为膀胱内灌注。膀胱内灌注也为适用于如本文所述的dHPG给药的一方法实例,所述dHPG给药用于作为预处理或联合治疗以增加组织药物摄取。膀胱内灌注为凭借将溶液插入至诸如膀胱的囊的一种药物递送手段。在递送至膀胱中,溶液通常借助插入的导管穿过尿道至膀胱给药。灌注溶液,通常停留在膀胱内一段时间,诸如约1小时或约2小时。灌注的通常容量为约10mL-约50mL的范围。停留时间后,将抽空溶液容量以及任何累积的已稀释了溶液的尿液以结束程序。停留时间代表膀胱内治疗期间最大的药物暴露时间,因为在抽空步骤期间除去了药物的大多数。其他的组合物或促进使组织递送局部化的方法的实例对本领域技术人员是明显的。例如,如本文所述的可使用的dHPG可在药物组合物中,其中dHPG在dHPG内核中包含紫杉烷或其他的疏水性药物,连同第二种药物,所述药物也可配制至HPG中,或第二种药物可与dHPG在溶液中组合用于递送。而且,dHPG可与靶向剂(例如,表皮生长因子受体的抗体,其在膀胱肿瘤、赫赛汀(Herceptin)或血管内皮生长因子(VEGF)中过表达)组合。
可通过本领域已知的方法配制适合的药物组合物并且通过技术人员确定它们的给药模式和剂量。对膀胱内灌注而言,掺入生物活性剂的dHPG可溶解于灌注媒介或缓冲系统以控制pH处于有利的水平,诸如约pH6-8,或另外适合的范围,例如约pH4-6或约pH8,所述灌注媒介诸如水、含水的共溶剂系统、诸如生理盐水或水中5%右旋糖的等渗的水溶液。可控制pH处于特定的范围以提供利于最佳化药物释放动力学、药物稳定性、最大的粘膜粘附、最大的溶解度或其组合。 也预期用于可溶于水的药物递送系统给药的其他药学上可接受的媒介。本领域技术人员已知的许多技术描述于Remington-The science and practice of pharmacy”(雷明顿-药物科学与实践),20th Edition(第20版),Jennaro等(Phila,Lippencott,Williams,and Wilkens,2000)。
如本文所用的药物组合物的“有效量”包括治疗上有效量或预防上有效量。“治疗上有效量”指的是剂量有效的并且持续必要的一段时间的量以获得理想的治疗结果,诸如减少癌细胞增殖、延长寿命或延长预期寿命。掺入生物活性剂的dHPG的治疗上有效量可根据因素而变化,诸如疾病状态、年龄、性别和个体的体重、以及生物活性剂引起个体内期望的反应的能力。可调整给药方案以提供最佳的治疗反应。治疗上有效量也为其中治疗上有利作用超过任何制剂的毒性或有害作用的量。“预防上有效量”指的是剂量有效的并且持续必要的一段时间的量以获得理想的预防结果,诸如预防或适应症进展的预防。通常在疾病之前或疾病的较早期,个体中使用预防剂量。
应注意的是剂量值可随着待被缓解的状况的严重性而变化。对任一特别的个体而言,特定的给药方案可根据个人需要和给药或监管组合物给药的人员的专业判断随时间调整。组合物的量可根据因素诸如疾病状态、年龄、性别和个体的体重而变化。可调整给药方案以提供最佳的治疗反应。例如,可快速灌注方式给药,可随时间分次剂量给药或剂量可如治疗情况的紧急状态所示的按比例地减少或增加。以剂量单位形式配制组合物有利于减轻剂量给药及一致性。
在一些实施方案中,如本文所述的dHPG可用于,例如并且不限于,与其他的治疗方法联合。例如,如本文所述的掺入生物活性剂的dHPG可与本领域技术人员已知的其他的治疗联合,作为新辅助的(之前)、附加的(期间),和/或辅助的(之后)治疗使用。
通常,如本文所述的dHPG可用于减少毒性。可利用标准的技术确定本文所述的dHPG的毒性,例如通过测试细胞培养物或实验动物以及确定治疗指数,即LD50(50%群体致死的剂量)与LD100(100%群体致死的剂量)间的比率。在某些情况下,然而,诸如在严重的疾病状况下,需要给予大量超额的组合物。在某些浓度时,本发明的一些dHPG为有毒的。滴定法研究可用于确定有毒的与无毒的浓度。通过检查特别的dHPG或组合物对细胞系的特异性评估毒性。动物研究也可用于提供聚合物是否具有对其他组织任何作用的指征。
可给予个体如本文所述的dHPG。如本文所用的,“个体”可为人类、非人类的灵长类动物、大鼠、小鼠、牛、马、绵羊、山羊、狗、猫等。可怀疑个体患有或处于患有癌症或其他的与具有粘膜表面的组织有关的疾病的危险中。此类适应症可为泌尿道(例如尿道和膀胱)、消化道(例如口、食道以及结肠)、气道(例如鼻和肺)、阴道腔和子宫颈以及腹膜腔。癌症(例如,膀胱癌、胃癌、食道癌、肺癌、喉癌、口腔癌、窦癌、阴道癌或子宫颈癌)、感染(例如,消化道或气道感染),和炎症性或自身免疫性疾病(例如,刺激性膀胱、炎症性肠疾病或慢性或急性炎症)以及其他的适应症可为本文所述的dHPG的理想的靶标,所述dHPG用于药物或其他的生物活性部分递送。用于癌症、感染以及炎症或性或自身免疫性疾病的诊断方法为本领域技术人员已知的那些。
例如,本文所述的dHPG可用于非肌层侵润性膀胱癌的治疗。本文所述的dHPG可用于药物制备,所述药物用于非肌层侵润性膀胱癌的治疗。本文所述的dHPG可用于治疗非肌层侵润性膀胱癌的方法中。所述方法包含给予需要其的个体有效量的本文所述的掺入生物活性剂(例如紫杉烷)的dHPG。例如,本文所述的dHPG可作为预处理或联合治疗使用以增加药物的药物摄取,所述药物用于非肌层侵润性膀胱癌的治疗。
参考已知的化学合成原则,本领域技术人员应理解制备或合成本文所述的dHPG的方法。例如WO2006/130978描述了适合的合成程序,所述程序可被认为是或者适当地将其调整以制备本文所述的聚合物。
用于dHPG的化学合成的通常的方法描述于下列非限制的示例性方案:
第二步,单体的添加,进一步地通过22-24小时内添加单体速率所定义。对凝聚的内核聚合物而言,烷基环氧化物的添加速率在反应期的早期较快而丙三醇环氧化物的添加速率在反应期的早期较慢。然而,不要求调整添加速率,只要组分的比率利于烷基组分在反应的早期的添加,相对于晚期。
本文描述了各种可选择的实施方案与实施例。这些实施方案与实施例为示例性并且不应理解为限制了本发明的范围。
实施例
实施例1:衍生的HPG的合成与特征
所有的化学药品均购于Sigma-Aldrich Canada Ltd(Oakville,Canada)并且没有进一步的纯化使用。所有的溶剂为来自于Fisher Scientific(Ottawa,Canada)的高效液相色谱(HPLC)等级并且没有进一步的纯化使用。
在配有机械搅拌器的三颈圆底烧瓶中进行聚合作用。第二个颈连接至双歧管史兰克线,以及第三个靠近穿过其添加试剂的橡胶隔片。典型的用于HPG-C8/10-MePEG聚合反应的程序如下。在氩气气氛下, 向烧瓶添加启动因子三羟甲基丙烷(TMP),然后添加含甲醇钾的甲醇溶液(20wt%)。利用磁性搅拌棒搅拌混合物15分钟,然后在真空中除去过量的甲醇。保持烧瓶在95℃下油浴中,并且利用注射泵在12小时内逐滴添加缩水甘油。单体添加结束后,再搅拌混合物5小时。然后添加辛基/癸基缩水甘油醚并且搅拌混合物24小时以形成HPG-C8/10。在12小时内逐滴添加MePEG350至该混合物然后再搅拌5小时。MePEG优选于PEG,因为MePEG的甲基保护单体的一端从而使得单体没有变成二价的,所述二价的可能导致dHPG分子间交联。也关注其他的保护基,包括合成后可以除去的那些。用这种方式,可与表面上的PEG链制备HPG,这可通过添加其他的化学基团或生物分子进一步地修饰,所述生物分子包括肽、糖肽、蛋白等。可修改本程序以合成不同的HPG,例如缩水甘油添加后可添加1,2-环氧十八烷至混合物以形成HPG-C18。
然后将产物溶解于甲醇并且通过将其穿过阳离子交换柱(AmberliteTMIRC-150)三次使其中和。通过己烷萃取除去未反应的辛基/癸基缩水甘油醚。除去甲醇,利用醋酸纤维素透析袋(MWCO:1000g/mol,Spectrum Laboratories Inc.),水作参比,伴随三次水变化/天,透析聚合物三天。然后通过冷冻干燥和热干燥获得干燥的聚合物。
可修改本程序以便合成凝聚的内核dHPG,其中烷基链为集中朝着聚合物的中心,代替常规的内核dHPG,其中遍及聚合物随机置放烷基链。通过以不同的速率和/或不同的比例将丙三醇环氧化物与烷基单体添加至反应混合物修饰聚合物的内核。为了形成凝聚的内核dHPG,在丙三醇环氧化物添加结束之前添加所有烷基单体,从而使得超支化结构的表面部分不包含任何烷基组分。反而烷基组分位于朝着HPG的内核。
通过首先制备如上文所述的HPG-C8/10合成HPG-C8/10-COOH。方案II显示了羧酸官能团的添加与HPG-C8/10的反应:
添加吡啶(50mL)至HPG-C8/10(0.5g)并且快速搅拌以溶解聚合物。添加二甲氨基吡啶(0.2g,0.0016摩尔),然后缓慢添加琥珀酸酐(12g,0.12摩尔)。室温下(大约22℃)搅拌过夜反应。添加水(100mL)并且搅拌混合物30分钟。通过旋转蒸发,伴随周期性添加水,除去溶剂,通过共沸蒸馏以使吡啶能够更好地蒸发。将残余物溶解于甲醇并且利用Spectra/Por透析膜(MWCO:3500g/mol),蒸馏水作参比,透析16小时。改变透析介质四次,每次用更高的甲醇浓度。透析介质的最终组合物为含70%甲醇的蒸馏水。通过旋转蒸发除去溶剂并且在真空烘箱中干燥聚合物过夜。
方案II显示了试图添加琥珀酰亚胺基碳酸酯至HPG-C8/10的第一反应方案。简单地说,真空、110℃下干燥HPG-C8/10然后冷却至室温。添加乙腈与DCM以溶解聚合物。然后添加N,N'-二琥珀酰亚胺基碳酸酯至烧瓶。排空烧瓶然后用氩气净化并且允许反应在添加吡啶后室温下过夜进行。反应后,通过旋转蒸发除去大多数乙腈。添加甲基叔丁基醚以沉淀聚合物。轻轻倒出上清液并且添加DCM以溶解聚合物。穿过10-15μm布氏漏斗(Buchner funnel)过滤物质以获得澄清的溶液,将其旋转蒸发除去DCM。添加MTBE以沉淀聚合物。真空室温下干燥最终的HPG-C8/10-NHS产物。
在显示了第一次尝试由此得到的高反应性、达到甚至储存在-20℃也不稳定,导致基质交联的程度的HPG-C8/10-NHS后,尝试第二合成途径。方案IV显示了尝试用于HPG-C8/10-NHS产生的第二反应方案。合成涉及产生如上文所述的作为中间体的HPG-C8/10-COOH,然后进一步地将其与NHS反应以产生HPG-C8/10-COOH-NHS。
HPG-C8/10与琥珀酸酐溶解于吡啶中并且在室温下反应24小时,二甲氨基吡啶(DMAP)作为催化剂。通过添加等量的水终止反应并且通过旋转蒸发溶液除去吡啶。透析HPG-C8/10-COOH的水溶液72小时(MWCO:3500g/mol)以除去残余的溶剂,并且冷冻干燥。进一步地在室温下将二甲基甲酰胺(DMF)中的HPG-C8/10-COOH与N-羟基琥珀酰胺(NHS)反应24小时,N,N'-二环己基碳二亚胺(DCC)作为催化剂。在反应的最后,通过旋转蒸发除去DMF。如上文所述的分离产物。用MTBE沉淀,乙腈中过滤,干燥之前用MTBE旋转蒸发及沉淀。
利用下列的,方案V中所总结的程序产生具有各种胺密度的HPG-C8/10-MePEG-NH2批次。各种试剂的化学计量学用于每一批次,表1中所述。
将HPG-C8/10-MePEG(4g)溶解于15ml无水1,4-二氧己环。用己烷漂洗氢化钾(0.45g)三次并且在真空下干燥。聚合物溶液与KH组合并且在室温下搅拌直至形成澄清的溶液,大约20%HPG-C8/10-MePEG上的OH基为去质子化的。通过溶解于二氯甲烷干燥N-(2,3-环氧丙基)邻苯二甲酰亚胺)(EPP)(1.184g)并且与Na2SO4或MgSO4搅拌过夜。过滤溶液并且在真空下干燥以除去二氯甲烷。将干燥的EPP溶解于无水1,4-二氧己环并且添加至聚合物,约85-90℃下搅拌过夜。通过将其穿过阳离子交换柱(AmberliteTMIRC-150)三次中和产物然后自乙醚沉淀三次以除去未反应的EPP。通过NMR,将15.5%苯邻二甲酰亚胺加入HPG-C8/10-MePEG。通过肼解作用实现苯邻二甲酰亚胺功能的剪切(用水合肼回流)。添加过量的水合肼溶液(2mL)至含聚合物的甲醇溶液并且将混合物回流48小时。回流后,蒸发甲醇,利用MWCO:1000g/mol膜透析48小时,水作参比并且冷冻干燥。
表1用于产生HPG-C8/10-MePEG-NH2的试剂化学计量学
通过NMR、FTIR、DSC以及TGA表征获得的聚合物。NMR特别适用于确定支链结构和添加至聚合物外壳的表面基团的存在。对一些表面化学而言,FTIR分析也用于确定羟基与它们被其他基团替代的消耗量,其中那些基团的化学提供了来自于其余的HPG结构的独特的红外光谱(IR spectrum),例如C=O键的添加。例如,FTIR可用于确定-COOH基团添加至表面或通过酯键的基团添加。
实施例2:紫杉醇或多西紫杉醇封装至dHPG
紫杉醇或多西紫杉醇连同dHPG可溶解于少量的乙腈中并且在60℃下烘箱内干燥1小时,然后用氮气流闪耀以清除有机溶剂的痕迹。由此得到的dHPG/紫杉醇或dHPG/多西紫杉醇基质可与10mM磷酸盐缓冲盐水(pH7.4)形成水合物,涡旋2分钟,60℃下烘箱内孵育1小时。由此得到的溶液通常为澄清的。在那些观察到白色沉淀的实例中,离心溶液(18000g,10分钟)并且将上清液转移至新的容器,保持在阴凉处直至使用。
实施例3:dHPG中多西紫杉醇与紫杉醇的稳定性
在DTX对失活的分解产物降解以及生物活性差向异构体(“7-表-DTX”)的互变现象方面表征掺入至dHPG的多西紫杉醇(“DTX”)的稳定性。已知紫杉醇与多西紫杉醇差向异构体的形成以均衡的形式出现而失活的分解产物的降解为不可逆的。利用超高效液相色谱(UPLC)分析各种dHPG中所述的稳定性。Waters Acquity UPLC BEH C18柱(2.1x50mm,1.7μm)用于主要降解峰的分离。注射量为3μL。流动相为10mM醋酸铵溶液,其通过称量0.385g盐并将它溶解于500mL HPLC等级的水制备。利用醋酸将pH调至pH4.0。在甲醇中制备DTX的储备溶液(2mg/mL)并且储存在-20℃冰箱。一组含DTX的标准液制备于在0.5-100μg/mL范围的50/50甲醇/水。检测限度(LOD)与定量限度(LOQ)为1μg/mL。1-100μg/mL的校正曲线与DTX的0.9998R2成线性关系。应用A 1/x加权。在LOQ(1μg/mL)与中距(10μg/mL)处验证方法准确度与精密度。在每个实例中,进行5次重复注射。表2总结了在DTX这些浓度处所获得的准确度与精密度。
表2通过UPLC检测DTX所获得的准确度与精密度
进行DTX的强迫降解以产生用于方法特异性评估的样品。通过制备含300μL的甲醇、150μL的5%氢氧化铵以及50μL的储备的DTX前体药物的溶液实现DTX的降解,所述DTX前体药物在碱性pH下几分钟内降解为DTX。监测DTX前体药物转化为DTX以及DTX降解两个多小时。2.2小时实验(室温)后,所有的前体药物有效地降解为DTX和其他的组分。利用上文所述的方法分析降解的样品以确定DTX前体药物、DTX以及相关的降解产物的分辨率。
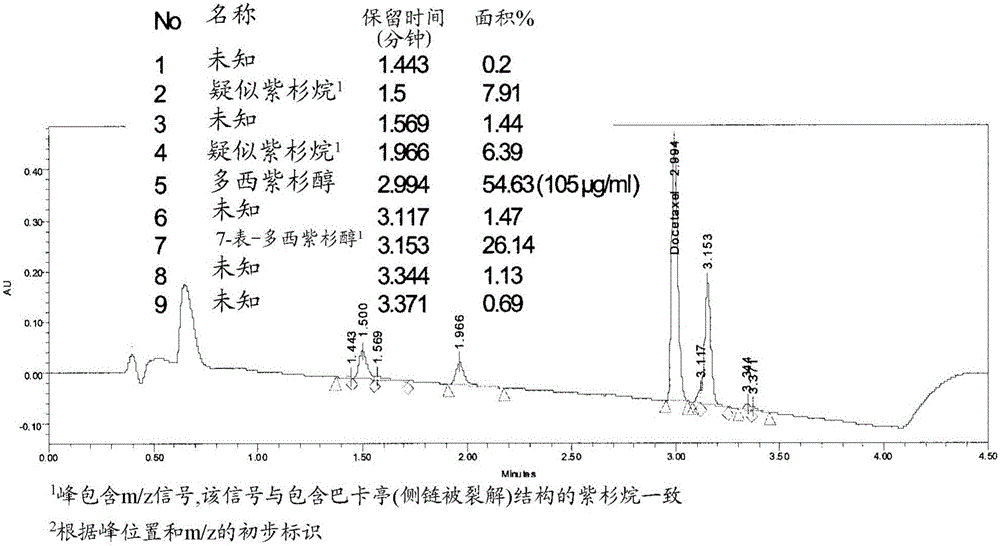
利用以上的UPLC方法,DTX为2.99分钟处的洗脱液并且7-表-DTX为3.15分钟处的洗脱液。样品色谱显示于图1中。1.4-2.0分钟的峰值为DTX与7-表-DTX的降解产物。根据时间计算DTX与7-表-DTX峰面积,然后用于确定残留在dHPG中DTX或表-DTX的百分比。结果显示于图2中。如图2中可见到的,当HPG聚合物为HPG-C8/10与HPG-C8/10-MePEG时,那些包含DTX的制剂为稳定的,所述制剂在缓冲至pH7.3PBS中,经过72小时,以DTX与7-表-DTX的形式保留超过90%掺入的DTX。药物的残余量已降至使组分失活并且通过质谱分析法MRM实验鉴定这些,这些的每个都贡献出多于2%的总样品数。这些实验旨在鉴定与DTX已知的降解产物有关的离子片段。
分别在图3A与3B中比较含DTX与7-表-DTX离子(+H+)m/z808.5的HPG-C8/10与HPG-C8/10-MePEG-NH2制剂的单一离子记录(SIR)。色谱图显示了7-表降解产物的存在,尽管它以更大的比例存在于HPG-MePEG-NH2样品中。对图3A与3B而言,最高的峰为DTX而较低的峰为7-表-DTX。尽管总离子色谱图由于制剂中聚合的成分显示了非常高的信号(图4),鉴定已知匹配DTX降解片段的附加质量(Kumar等,2007,多西紫杉醇药物物质中降解杂质及其制剂的分离与特征,Kumar等,Journal of Pharmaceutical and Biomedical Analysis 12 March 2007 43(4):1228-1235),与最大的降解产物峰一致。在1.5分钟处观察到226与282的M/z,这对应来自DTX侧链的片段。然而,存在583的m/z,这对应10-脱乙酰巴卡亭III+K+,表明紫杉烷包含内核与侧链二者。也观察到320与562的M/z。2分钟处的峰具有m/z=581和583,这可分别对应10-氧-10-脱乙酰巴卡亭III+K+和10-脱乙酰巴卡亭III+K+。该峰值位置也在其中期望观察到巴卡亭降解产物的色谱图区域内。
可通过调节制剂的pH进一步地增加一些制剂的稳定性。例如,含掺入DTX的HPG-C8/10-MePEG-NH2的组合物,按比例与缓冲盐溶解于水介质以便获得pH5.5-6.5,比相同的不包括缓冲盐的组合物或具有改变的缓冲盐组合物的组合物,例如产生pH7.4的PBS缓冲液,更稳定。适当的缓冲盐包括磷酸盐缓冲盐。可选择地,可通过添加诸如HCl的酸至组合物降低组合物的pH。而且,通过改变组合物的pH显著减慢DTX的降解。
实施例4:dHPG的体外生物相容性
通过检测不同的dHPG制剂是否能够杀死KU7癌细胞测量dHPG的毒性。利用没有掺入任何药物或生物活性部分的dHPG进行这些实验。结果显示于图7A与7B中。图7A显示了KU7细胞增殖的百分比与HPG-C8/10的浓度的函数,对常规的内核制剂和凝聚的内核制剂二者而言。改变内核体系结构不会显著地影响HPG-C8/10的稳定性。图7B显示了KU-7细胞的活力与HPG-C8/10-MePEG的浓度的函数,对常规的内核制剂和凝聚的内核制剂二者而言。与常规的内核聚合物相比,具有凝聚的内核聚合物显示了高于细胞活力IC50 10倍。具有凝聚内核的dHPG对细胞的耐受性好于具有常规内核的dHPG。凝聚的内核制剂对KU-7细胞的毒性比常规的内核制剂更小。表3显示了用于制备图7A与7B中所示制剂的单体的体积比。
表3用于制备图7A与7B中所示制剂的单体的体积比
图8显示了无MePEG外壳的HPG-C8/10聚合物为其中耐受性最小的,并且无MePEG外壳,自常规的内核改变为凝聚的内核体系结构在改善细胞活力方面没有益处。HPG-OH表示它为表面上无MePEG的HPG。然而,当添加MePEG时,益处变得显而易见的。HPG-C8/10-MePEG6.5通式(具有6.5mol MePEG/HPG)显示比得上HPG-C8/10的耐受性,当比较正常的内核版本时,但是当使用凝聚的内核体系结构时,HPG-C8/10-MePEG6.5的耐受性改善了常规的内核HPG-C8/10-MePEG13的水平,其使外壳中的MePEG的量加倍。早先公开的HPG-MePEG聚合物(Mugabe C等,2008BJUI 103:978-986)也具有更高的MePEG量,尽管没有对它们定量,估计它们>15mol%并且耐受良好。
实施例5:体外细胞摄取测定显示dHPG被运输至细胞内
检查KU7细胞对荧光素标记的HPG-C8/10-COOH-NHS与HPG-C8/10-COOH的摄取。以1mg/mL将dHPG溶解于无胎牛血清(FBS)基质中。在12孔平板的每个平板中,将250μL dHPG暴露于盖玻片上的细胞。用PBS洗涤平板两次然后用含3.7%甲醛的PBS固定10分钟。再用PBS洗涤平板两次。利用具有苯基吲哚(DAPI)Prolong GoldTM固定盖玻片。
也观察HPG-C8/10-COOH的Z电势以便确定聚合物实际上进入了KU-7细胞并且不只是留在细胞表面。因为以所有的观察角度观察荧光,所以发现dHPG在细胞内部。这些结果显示dHPG进入了细胞并且未引起细胞内任何不良反应。体外数据显示,当HPG暴露于细胞时,HPG-C8/10-COOH-NHS与HPG-C8/10-COOH以1小时进入。然而,在体内,接触不如体外方案完全,要求延长暴露时间以促进这一摄取。
实施例6:紫杉烷负载至凝聚的内核和常规的内核dHPG
研究凝聚的与常规的内核HPG-C8/10-MePEG的最大药物负载。将DTX与PTX负载至HPG-C8/10-MePEG以靶向0.5,1.0,2.0及3.0mg/mL的药物浓度。制备含100mg/mL聚合物的四氢呋喃(THF)溶液并且添加DTX或PTX。N2流下干燥THF约2小时,然后在排风罩中干燥过夜。HPG-C8/10-MePEG/PTX和HPG-C8/10-MePEG/DTX基质与PBS缓冲液(pH 7.4)形成水合物。以14000转速旋转减慢由此得到的溶液约15分钟。通过HPLC检测上层清液的液体以获得封入HPG-C8/10-MePEG的药物的浓聚物。结果显示于表4中。
表4DTX与PTX至凝聚的内核(“CC”)和常规的内核(“RC”)HPG-C8/10-MePEG的理论与实际负载
发现DTX负载优于PTX。
实例例7:HPG-C8/10与HPG-C8/10-MePEG的合成及特征
根据报道的方案(Kainthan,R.K.,Mugabe,C.,Burt,H.M.,Brooks, D.E.,2008.Biomacromolecules 9,886-895),用基于环氧化物的开环聚合反应的单一的锅合成程序进行辛基/癸基缩水甘油醚(O/DGE,C8/10)内核修饰的HPG的聚合作用。
所有化学品均购于Sigma-Aldrich(Oakville,ON)并且所有溶剂为来自Fisher Scientific(Ottawa,ON)的HPLC等级。从MePEG 350、氢氧化钠以及环氧氯丙烷的反应合成α-环氧树脂、ω-甲氧基聚乙二醇350(MePEG 350环氧化物)。从Sigma–Aldrich获得辛基/癸基缩水甘油醚、甲醇钾以及三羟甲基丙烷(TMP)并且没有进一步纯化使用。
将120mg启动因子(TMP)与1.5ml甲醇钾溶液混合在甲醇(25%,w/v)中并且在氩气下添加至三颈圆底烧瓶。在105℃下搅拌混合物1小时,在之后真空下除去过量的甲醇,然后,利用注射泵,以1.4ml/小时的速率将13ml缩水甘油与9ml O/DGE混合物注射至启动因子。利用数字化头顶搅拌系统(BDC2002)将搅拌速率固定在68转速。单体添加完成后,再将混合物搅拌6小时。通过己烷萃取以除去未反应的辛基/癸基缩水甘油醚获得纯化的聚合物。然后将产物溶解于甲醇中并且通过穿过阳离子交换柱(Amberlite IRC-150,Rohmand Haas Co.,Philadelphia,PA)三次中和。真空下除去甲醇然后利用醋酸纤维素透析袋(MWCO 10,000g/mol,Spectrum Laboratories),水作参比,透析聚合物的水溶液三天,换水三次/天。
1HNMR(400MHz,D6-DMSO)δH:0.75-0.82(-CH3,TMP);0.82-0.91(O/DGE上-CH3-烷基);1.16-1.53(-CH2-,O/DGE上烷基);2.46(溶剂,D6-DMSO);3.16-3.80(-CH与-CH2-,来自HPG内核);4.8(-OH)。
制备含不同MePEG量的HPG-C8/10-MePEG并且指定HPG-C8/10-MePEG6.5与HPG-C8/10-MePEG13表示添加供给的MePEG量(分别为6.5与13mol MePEG/HPG摩尔)。以如HPG-C8/10反应类似的方式进行合成,除了在合成的最终步骤中添加不同的MePEG量至反应混合物。烷基(R)衍生的HPG-C8/10-MePEG的一锅合成反应图解总结于方案VI中。
将120mg启动因子(TMP)与1.5ml甲醇钾溶液混合在甲醇(25%,w/v)中并且在氩气下添加至三颈圆底烧瓶。在105℃下搅拌混合物1小时,在之后真空下除去过量的甲醇,然后,利用注射泵,以1.4ml/小时的速率将13ml缩水甘油与9ml O/DGE混合物注射至启动因子。所有缩水甘油与O/DGE的混合物注射后,反应继续约6小时。然后添加0.1ml氢化钾(KH)至烧瓶。搅拌混合物1小时,之后利用注射泵,以1.4ml/小时的速率,添加10ml或20ml MePEG 350环氧化物作为“一锅”合成法中的最终步骤。根据HPG上的靶标密度添加MePEG 350量(即10ml MePEG 350支持每摩尔HPG上6.5mol MePEG靶标)。然后增加搅拌速率至90转速并且在105℃下连续地进行反应过夜。通过己烷萃取除去未反应的辛基/癸基缩水甘油醚的任何痕迹。产物溶解于甲醇中并且通过穿过阳离子交换柱(Amberlite IRC-150,Rohm与Haas Co.,Philadelphia,PA)三次中和。真空下除去甲醇然后利用醋酸纤维素透析袋(MWCO 10,000g/mol,Spectrum Laboratories),水作参比,透析聚合物的水溶液三天,换水三次/天以除去未反应的MePEG环氧化物。然后通过冷冻干燥获得干燥的聚合物。
1HNMR(400MHz,D6-DMSO)δH:0.75-0.82(-CH3,TMP);0.82-0.92(O/DGE上-CH3-烷基);1.15-1.55(-CH2-,O/DGE上烷基);2.50(溶剂,D6-DMSO);3.15-3.80(-CH与-CH2-,来自HPG内核);3.23(-OCH3-来自MePEG),3.32(残留水);4.8(-OH)。
利用甲醇钾通过来自于部分去质子化的三羟甲基丙烷(TMP)的缩水甘油的阴离子开环多支链聚合作用制备HPG-C8/10(无MePEG链的HPG)。HPG-C8/10具有许多的末端羟端基,数量/分子大致等于聚合度。 用C8/10烷基链衍生HPG-C8/10内核以产生疏水性内核,以允许药物负载,例如紫杉烷。MePEG链与HPG上的羟基连接。因为在其他组分反应后将MePEG 350环氧化物添加至聚合反应,形成了亲水性外壳以增加HPG的水溶解度。
NMR实验旨在表征HPG聚合物的结构。利用氘化溶剂,通过Bruker Avance 400MHz NMR光谱仪上所记录的异核单量子相关谱(HSQC)NMR实验(Cambridge Isotope Laboratories,99.8%D)估算HPG上的MePEG与烷基链的分数。残留溶剂峰引用化学位移。利用Sparky(T.D.Goddard and D.G.Kneller,Sparky 3,University of California,San Francisco)分析HSQC光谱。图9和10显示了HPG-C8/10与HPG-C8/10-MePEG聚合物的代表性质子与2维HSQC光谱。将所有峰分配给HPG的结构组分,利用原料光谱作为起始参考(图10B)。质子NMR光谱类似于所报道的那些(Kainthan,R.K.,Janzen,J.,Kizhakkedathu,J.N.,Devine,D.V.,Brooks,D.E.,2008.Biomaterials 29,1693–1704;Kainthan,R.K.,Mugabe,C.,Burt,H.M.,Brooks,D.E.,2008.Biomacromolecules 9,886–895)。HSQC NMR数据确认HPG结构具有光谱中明显的分支体系结构的超支化聚合物(图9和10)。通过HSQC实验中体积积分计算每一取代基的分数。通过比较MePEG甲氧基与O/DGE甲基的积分和TMP CH3基积分,计算对每一HPG聚合物而言的O/DGE与MePEG(mol/mol)的分数。HSQC数据显示了未反应的环氧化物单体的缺失(图10),这表明通过未反应的单体聚合物污染的缺失。
通过具有多角度激光散射检测的凝胶渗透色谱(GPC-MALLS)确定dHPG聚合物的分子量与多分散性。分子量为约80,000g/mol(表5)。
dHPG的物理化学特征总结于表5中。
表5负载PTX与DTX的HPG-C8/10-OH与HPG-C8/10-MePEG的物理特征
1Tg,取自转化正中心的玻璃化转变。
2Td,取自最大重量损失的降解温度。
3PTX与DTX在HPG-C8/10-MePEG的最大负载能力时负载。
Mw,通过与MALIS检测仪连接的凝胶渗透色谱法(GPC-MALLS)所确定的重量平均分子量。
Mw/Mn多分散性。
ND,未确定的。
实施例8:纯化方法对dHPG的热力特性的影响以及对负载PTX与DTX的dHPG的物理及化学稳定性的影响
通过差示扫描量热法(DSC)和热重量分析(TGA)评估纯化方法对dHPG的热力及降解特性的影响。将已经过各种纯化步骤的聚合物称为纯化的dHPG,所述纯化步骤,例如己烷萃取以除去未反应的C8/10烷基链,随后通过阳离子交换柱中和,然后透析。
利用TA仪器DSC Q100及TGA Q50进行热力分析。通过经过以10℃/分,-90℃-85℃温度范围内的“热-冷-热”循环使在气密封口铝锅中的称出试样循环,获得DSC产量。以恒定的升温程序(20.0℃/分-500℃/分),40ml/min的气流量(氮气)进行TGA产量。记录实时重量百分比和TGA室温。利用TA通用分析2000软件(TA Universal Analysis 2000software)(4.2E版本,TA仪器)进行数据分析以发现起始点。利用配备AB104-S平衡的Mettler Toledo DL39Karl Fisher电量计, 通过滴定法确定dHPG的水含量。将已知量的dHPG溶解于无水甲醇中并且用试剂(Sigma)滴定。通过减去来自于无水甲醇的背景读数获得最终的结果。
随着MePEG含量自0(HPG-C8/10)下降至4.6mol MePEG/HPG(表5),在温度自-38℃下降至-55℃时,HPG-C8/10与HPG-C8/10-MePEG表现出玻璃化转变。观察到纯化方法对dHPG的Tg值没有影响并且没有观察到纯化方法对热稳定性的显著影响。纯化的与未纯化的dHPG二者在温度高达300℃时为稳定的,没有热分解的迹象(表6)。
也评估纯化方法对负载PTX与DTX的HPG-C8/10-MePEG的物理及化学稳定性的影响。PTX与DTX二者均负载至纯化的或未纯化的HPG-C8/10-MePEG并且通过观察药物自PBS(pH 7.4)中沉淀的起始评估物理稳定性。通过LC/MS/MS评估化学稳定性以确定PTX与DTX的量以及它们的降解产物。
纯化方法影响负载PTX与DTX的dHPG的物理及化学稳定性。对未纯化的聚合物而言,最大可获得的PTX与DTX负载量更大并且发现这些制剂比用纯化的HPG所制备的制剂的物理性更稳定(表6)。未纯化的聚合物所制备的样品在几天内(>3天)未沉淀,紫杉烷负载高达5%(w/w)而纯化的聚合物所制备的那些及5%(w/w)紫杉烷负载在几小时内沉淀或就在PBS中组成(表6)。然而,观察到PTX与DTX在未纯化的dHPG中化学性不稳定并且在制剂制备期间大部分紫杉烷降解。约75-80%PTX紧跟着负载在未纯化的HPG-C8/10-MePEG中就降解,不管其是否为干燥的(聚集的)基质或是否在其在PBS(pH 7.4)中重组后(表6)。然而,一旦缓冲液中无进一步的PTX降解发生,相反,干燥的基质中的PTX就继续降解24小时。图11B显示了来自未纯化的HPG-C8/10-MePEG中PTX制剂的具有对应于PTX的峰及源自几个降解产物形成的其他峰的色谱图。自干燥溶剂后(例如聚集状态下,在PBS中组成之前)就溶解于乙腈的制剂中获得色谱图。通过分别具有587及854m/z值的LC/MS/MS鉴定两个主要的降解产物。基于这些质量及这些峰的相对保留时间,假设它们的身份分别为巴卡亭III(m/z587,图11A)和7-表-紫杉酚(m/z 854)。未纯化的制剂中也观察到其他的 降解产物(峰A&B,图11B)。基于它们的相对保留时间,峰面积以及紫杉烷的已知降解机制,据信峰B(图11B)为巴卡亭V,其为7-表-巴卡亭III,而据信峰A为10-脱乙酰-巴卡亭III。然而负载在纯化的聚合物中的紫杉烷,展现了不同的行为。发现PTX在聚集及溶液中几天内化学性稳定并且在制剂制备期间没有观察到主要的降解产物(表6和图12C)。观察到PTX与DTX在未纯化的HPG中快速降解并且据信这归因于未纯化的聚合物中碱性杂质的存在。最可能的基本杂质来自于过量的甲醇钾及HPG合成期间所添加的氢化钾。甲醇钾与氢化钾二者为强碱并且与聚合物中残留水分结合将创造利于C7位置上的差向异构作用和PTX的酯剪切的环境以产生巴卡亭III(或对DTX而言,10-脱乙酰-巴卡亭III)(图11A)。dHPG聚合物在蒸馏水中的pH的测量显示未纯化的聚合物具有碱性pH而纯化的聚合物具有酸性pH(由于用Amberlite IRC-150,阳离子交换树脂处理),对紫杉烷而言更稳定的环境。已报道了紫杉烷的最大稳定性为在3-5pH范围内(Dordunoo,S.K.,Burt,H.M.,1996.Int.J.Pharm.133,191-201;Tian,J.,Stella,V.J.,2010.J.Pharm.Sci.99,1288-1298)。纯化的聚合物在这一pH范围内(表6),因此改善了负载的紫杉烷的化学稳定性。通过负载药物的大多数降解为比母体紫杉烷更小的并且更加亲水的分子(巴卡亭III与巴卡亭V)的事实解释负载在未纯化的dHPG聚合物中的紫杉烷具有明显更高的药物负载量及更大的化学稳定性,提示这些降解的分子更有效地负载至dHPG中。
表6聚合物纯化对聚合物特性的影响,以及对纯化的及未纯化的HPG-C8/10-MePEG13所制备的负载紫杉烷的制剂的物理与化学稳定性的影响
HPG-C8/10-MePEG13中1PTX与DTX负载量(%,w/w),也组成等价的水浓度(mg/ml)。
2“聚集的”基质表示通过在与PBS缓冲液组成之前溶剂蒸发制备的负载紫杉烷的HPG-C8/10-MePEG13聚合物。对聚集的基质而言t=0为就在其最终的制备步骤之后,干燥以除去溶剂。
实施例9:MePEG衍生作用对dHPG的热力特性、表面电荷以及颗粒大小的影响
利用Malvern NanoZS颗粒大小分析仪进行颗粒大小与Z电势分析,每个分析使用DTS0012一次性定径比色皿。在1mM NaCl中制备浓度15mg/ml的聚合物溶液并且用0.22μm注射器式滤器(PALLAcrodisc具有13mm尼龙膜)过滤。样品采集参数为:角度为具有自动衰减的173°反散射;游程数11(10s/游程);分散剂为25℃水(粘性0.8872cP,阻力指数(RI)1.330);Mark-Houwink参数A=0.428及K=7.67e-05cm2/s。假定dHPG具有与RI=1.460及0.01吸收的聚乙二醇(PEG)相似的折射率。最终的数据代表所有游程的平均值。
具有PTX与DTX负载的dHPG颗粒大小始终直径小于10nm, 所述PTX与DTX负载对HPG-MePEG无影响(未显示数据)。负载HPG的药物形成小于10nm的非常小的纳米颗粒。
HPG表面上MePEG链的存在对这些纳米颗粒上的总体表面电荷没有显著的影响,如通过Z电势所测量的如下:HPG-C8/10=-1.29±0.97mV;HPG-C8/10-MePEG6.5=-0.92±1.68mV;HPG-C8/10-MePEG13=0.18±0.16mV。
观察到随着递增的MePEG密度,对玻璃化转变温度的影响。分别对HPG-C8/10聚合物而言,Tg自-37.5℃下降至-45.2℃,对HPG-C8/10-MePEG6.5与HPG-C8/10-MePEG13而言,Tg自-37.5℃下降至-55.4℃(表5)。PTX或DTX负载也使Tg下降(表5)。
实施例10:dHPG中PTX与DTX的负载量、量化和稳定性以及来自于dHPG的PTX与DTX的释放
将PTX或DTX与dHPG溶解于含1ml乙腈溶液的4ml小瓶中并且在烘箱中60℃下干燥1小时,氮气闪耀以清除有机溶剂的痕迹。由此得到的dHPG/紫杉烷基质与1ml 50℃下温热的10mM磷酸盐缓冲盐水(PBS,pH7.4)形成水合物并且涡旋2分钟。由此得到的溶液通常为澄清的但是如果在其中观察到白色颗粒,离心溶液(18,000×g 10分钟)并且将上清液转移到新的小瓶。
通过如早先所述的反相HPLC确定PTX与DTX掺入至HPG的量(Jackson,J.K.,Smith,J.,Letchford,K.,Babiuk,K.A.,Machan,L.,Signore,P.,Hunter,W.L.,Wang,K.,Burt,H.M.,2004.Int.J.Pharma.283,97-109)。将100μl dHPG/PTX或DTX溶液与900μl乙腈/水(60:40,v/v)溶解并且转移至HPLC小瓶(Canadian Life Science,Peterborough,ON)。利用具有含乙腈、水以及甲醇(58:37:5,v/v/v)混合物的流动相对称性C18柱(Waters Nova-Pak,Milford,MA),以1ml/min的流速进行药物含量分析。样品注射体积为20μl并且利用处于232nm波长的UV检测进行检测。HPG-C8/10具有有限的水溶性,导致紫杉烷的低药物负载量(未显示数据)。dHPG中烷基(C8/10)链的存在对疏水性药物的负载是重要的,然而它也显著地降低了它们的水溶性。为增加HPG的水溶 性,在这些分子合成期间,反应的末期添加MePEG 350链。dHPG中MePEG量的相对少的增加导致HPG-C8/10-MePEG13增加的药物负载量,对PTX与DTX二者而言。HPG中DTX负载量高于PTX的负载量。负载DTX的dHPG显示了比PTX制剂更大的物理稳定性。HPG-C8/10-MePEG13中DTX的最大负载量(5%,w/w)高于PTX的最大负载量(2%,w/w)。
评估负载紫杉烷的dHPG的物理与化学稳定性。通过目视观测制剂的澄清度评估物理稳定性,小于24小时内的沉淀被认为物理学上不稳定的制剂。PBS中再水合一发生就观察样品(t=0),或室温下1,3,6,24,48以及72小时后。通过上文所述的HPLC方法监测PTX与DTX的化学稳定性。通过质谱分析法,利用Waters TQD质谱仪鉴定降解产物。电喷射离子源阻断温度150℃,脱溶剂温度350℃,采样锥电压45kV,毛细管电压0.70kV,提取器电压3kV,RF电压0.1kV,进样锥气体流速25l/h,脱溶剂气体流速600l/h以及碰撞气体流速0.2ml/min。分子经受阳离子模式的电喷雾离子阱。
通过透析方法确定HPG释放的PTX与DTX。称量100mg dHPG(HPG-C8/10-MePEG6.5或HPG-C8/10-MePEG13)并且1mg PTX或DTX在1ml乙腈溶液中混合,用15uCi 3H-DTX或3H-PTX(15μl)脉冲并且在氮气流下干燥以除去溶剂。自Moravek Biochemicals and Radiochemicals(Brea,CA)获得放射性药物(3H-DTX或3H-PTX)。dHPG/紫杉烷基质与2ml PBS形成水合物并且转移至透析袋,以500ml人造尿液(pH 4.5或6.5)作参比,100转速震动透析。透析膜管购于Spectrum Laboratories(Rancho Dominguez,CA)。根据Brooks等的方法(Brooks,T.,Keevil,C.W.,1997.Lett.Appl.Microbiol.24,203–206)制备人造尿液,未添加蛋白胨或酵母提取物。利用0.1M HCl将溶液的pH调至pH4.5和/或6.5。在不同的时间点,测量透析袋的体积并且取10μl样品用于透析袋中残余的放射性的测量并且将全部的对外释放介质与新鲜的介质交换以维持下沉状态。通过β闪烁计数器(Beckman Coulter Canada,Mississagua,ON)确定每一时间点透析袋中残留的3H-DTX或 3H-PTX的浓度。通过减去来自实验开始时药物的初始量的每一时间点 残留的药物的量计算所释放药物的累积百分比。数据表达成根据时间所释放的药物的累积百分比。数据代表三个独立的实验的平均值(SD)。
尿液的pH通常为酸性的但是已知在宽的范围(pH 4.5-8)内变化,因此评估pH对负载在HPG-C8/10-MePEG中PTX与DTX释放度的影响。通过连续控制释放与很少或无释放的突发相以及随后较慢的缓释相表征来自于HPG的紫杉烷的释放度。自HPG-C8/10-MePEG释放DTX比PTX迅速(2天后75%对50%药物释放)几乎所有DTX释放在6-7天,与对PTX而言12-14天相比。据信这由于DTX更大的疏水性而更疏水的PTX可具有更大的相容性并且与HPG内核的烷基链(C8/C10)相互作用导致较慢的药物释放速率。观察到HPG上MePEG密度的增加对药物释放没有影响(图13A)。观察到释放介质的pH变化(pH4.5-6.5)对来自于HPG-C8/10-MePEG纳米颗粒的药物释放没有影响(图13B)。利用各种动力学模型(包括一阶,higuchi以及korsmeyer模型)来自于HPG纳米颗粒的PTX与DTX释放度的评估表明一阶与higuchi动力学二者提供了最佳匹配r2=0.98-0.99(数据未显示)。在药物释放速率方面,制剂间没有统计学差异(数据未显示)。
实施例11:HPG-C8/10-MePEG13的若丹明标记与若丹明标记的HPG-C8/10-MePEG13细胞摄取
根据Huang等的方法,稍加改动(Huang,S.N.,Phelps,M.A.,Swaan,P.W.,2003.J.Pharmacol.Exp.Ther.306,681-687),将四甲基若丹明-5-羧基叠氮化物(TMRCA)共价标记于HPG-C8/10-MePEG13。将500mg HPG-C8/10-MePEG13溶解于5ml无水1,4-二氧己环中。将适当量的TMRCA溶解于无水1,4-二氧己环以产生1mg/ml的最终浓度。将对应于大约20mol%HPG的这一荧光探针的675μl等分试样添加至HPG-C8/10-MePEG13溶液并且在80℃下、油浴中,氮气流下加热搅拌5小时。以二甲基甲酰胺(DMF)(MWCO 12,000-14,000)作参比,透析溶液直至透析液为无色的,然后以蒸馏水作参比,透析24小时。冷冻干燥荧光标记的聚合物(HPG-C8/10-MePEG13-TMRCA)并且储存在-80℃下琥珀色小瓶中。
允许KU7细胞生长在几个位于10cm有盖培养皿的底部的显微镜1cm×1cm盖玻片上,直到达到~75%细胞覆盖,其对应于大约7×104个细胞的细胞数量。用温热的PBS洗涤这些包含细胞的盖玻片三次然后将其置于保鲜膜衬里的有盖培养皿,细胞侧朝上。将250μl HPG-C8/10-MePEG13-TMRCA溶液(1mg/ml溶解于改良培养基(DMEM))添加至盖玻片。细胞与HPG-C8/10MePEG13-TMRCA孵育1,4,8及24小时。为对照,将KU7细胞孵育于没有任何补充的DMEM中。然后用PBS缓冲液用力地洗涤盖玻片四次,轻轻地吸干过量的PBS,添加250μl 3.7%多聚甲醛以固定细胞10分钟。用PBS再洗涤盖玻片三次并且沉入水中。吸干过量的液体后,用具有苯基吲哚(DAPI)的 不变色的试剂(分子探针,Invitrogen)将细胞染色并且在显微镜载玻片上,玻片装载的细胞侧朝下。通过无色指甲油密封盖玻片的边缘以避免干燥。黑暗中孵育样品过夜以确保细胞的合适染色。在配备有DAPI(λex 340-380nm;λem,435-485nm;二向色分离器,400nm)与若丹明(λex 530-560nm;λem,590-650nm;二向色分离器,570nm)滤器的Olympus FV-1000倒置共聚焦显微镜下观察样品。进行直接对比(DIC)以显现细胞膜并且用405nm激光激活。为了清楚地显示标记的聚合物在细胞内部,通过荧光与DIC分析图像。
通过KU7细胞的共聚焦显微镜显现若丹明标记的HPG-C8/10-MePEG13(HPG-C8/10-MePEG13-TMRCA)。孵育1小时后,存在HPG-C8/10-MePEG13-TMRCA摄取至KU7细胞的证据。HPG-C8/10-MePEG13-TMRCA在1h时摄取的代表性图像显示于图14中。面板A显示了DAPI染色的未处理的KU7细胞,其允许细胞核以蓝色显现(图像中显示为白色)。图像为直接对比信号(其显示了细胞的轮廓)与荧光信号(其显示细胞核(白色)以及任何其他的荧光缺失)的叠加。面板B显示已与HPG-C8/10-MePEG13-TMRCA纳米颗粒孵育了1小时的KU7细胞。通过细胞核(DAPI染色的蓝色)周围聚合物的红色荧光(红色荧光在图像中显示为白色部分。细胞核周围的红色荧光在图像中显示为深色部分)显示HPG-C8/10-MePEG13-TMRCA在细胞浆中的存在。来自于面板B的相同细胞群体的堆叠(未显示堆叠)证明了红色 荧光的纳米颗粒存在遍及细胞浆,而不是仅仅粘附或存在于细胞膜。这些纳米颗粒似乎均匀地分布于细胞浆中,尽管观察到一些点状结构,所述点状结构表明HPG-C8/10-MePEG13-TMRCA被包装至用于细胞运输的小媒介中。KU7细胞的细胞核腔室内没有检测到来自聚合物的荧光。通过1小时的孵育,KU7细胞吸收HPG-C8/10-MePEG13-TMRCA纳米颗粒并且在1,4,8或24小时时间点所获得的图像中没有差别。
实施例12:HPG-C8/10-MePEG的体外细胞毒性研究
根据如上文所述的报道的方案(Kainthan RK,Mugabe C,Burt HM,Brooks DE.Biomacromolecules,9(3),886-895(2008)),制备HPG-C8/10-MePEG。1H NMR(400MHz,D6-DMSO)δH:0.75-0.82(-CH3,TMP);0.82-0.92(-CH3-烷基在O/DGE上);1.15-1.55(-CH2-,烷基在O/DGE上);2.50(溶剂,D6-DMSO);3.15-3.80(-CH与-CH2-,来自HPG内核);3.23(-OCH3-来自MePEG),3.32(残留水);4.8(-OH)。
通过异核单量子相关谱(HSQC)NMR实验估算MePEG与HPG上的烷基链的分数。化学位移参考残余的溶剂峰。通过凝胶渗透色谱法与多角度激光散射仪(GPC-MALLS)确定聚合物的分子量与多分散性:分子量=83,000g/mol,多分散性1.22(数据未显示)。
利用Malvern NanoZS颗粒大小分析仪进行颗粒大小分析。负载HPG-C8/10-MePEG药物形成小于10nm的纳米颗粒(7.5±3.4nm-7.8±2.7nm,数据未显示)。
通过将PTX(1mg)或DTX(0.5mg)与HPG-C8/10-MePEG(100mg)溶解于4ml小瓶中的1ml乙腈溶液中并且在60℃下烘箱中干燥1小时以及氮气流闪耀以清除有机溶剂的痕迹,制备负载的dHPG的PTX或DTX。紫杉醇(PTX)粉末获取自Polymed Therapeutics,Inc.(Houston,TX)。多西紫杉醇(DTX)粉末获取自Natural Pharmaceuticals Inc.(Beverly,MA)。由此得到的HPG-C8/10-MePEG/紫杉烷基质与1ml10mM磷酸盐缓冲盐水(PBS,pH 6)形成水合物并且涡旋2分钟。通过反相HPLC确定掺入至HPG-C8/10-MePEG的PTX与DTX的量。通过溶剂蒸发方法,PTX与DTX可以高药物负载量(最大负载量分别为2和5%w/w)负载。
评估商品化制剂与以及负载PTX和/或DTX的HPG-C8/10-MePEG制剂针对KU7-荧光素酶细胞系、低级的(RT4,MGHU3)与高级的(UMUC3)人尿路上皮癌细胞系的细胞毒性。来自于Bristol-Myers-Squibb(Princeton,NJ)。来自于Sanofi-Aventis Canada Inc.(Laval,Quebec)。人膀胱癌细胞系RT4与UMUC3购于美国典型培养物保藏中心(American Type Culture Collection)。保持细胞在含10%热灭活的胎牛血清的McCoy’s介质(Invitrogen,Burlington,ON)中并且保持在37℃下,湿润的5%CO2大气中。作为慷慨的礼物,MGHU3细胞获取自Dr.Y.Fradet(L’Hotel-Dieu de Quebec,Quebec,Canada)并且保持在补充了10%胎牛血清与2mM左旋谷氨酰胺的MEM(Invitrogen)中。Dr.C.Dinney(MD Anderson Cancer Center,Houston,TX,USA)友好地提供了KU7并且保持在含5%胎牛血清的DMEM中。出于显示的目的,通过Dr.Graig Logsdon(M.D.Anderson Cancer Center,Houston,TX,USA)用含萤火虫荧光素酶基因的慢病毒感染KU7细胞,并且这些亚克隆被命名为如早先报道的KU7-荧光素酶(Hadaschik BA,Black PC,Sea JC等.BJU Int,100(6),1377-1384(2007))。细胞以5,000个细胞/孔沉积于100μl体积的补充了10%FBS的McCoy’s介质96孔平板中并且在添加新鲜制备的负载PTX的HPG-C8/10-MePEG或负载DTX的HPG-C8/10-MePEG溶液前允许平衡24小时。细胞暴露于药物制剂2小时以模拟目前的用于灌注治疗的临床标准并且如早先报道的(Hadaschik BA,ter Borg MG,Jackson J等,BJU Int,101(11),1347-1355(2008)),利用CellTiter96水的非放射性细胞增殖测定(CellTiter96Non-Radioactive Cell Proliferation)(MTS)(Promega,Madison,WI)在72小时后确定细胞活力。每一实验重复三次并且MTS值适合于所有细胞系的线性吸光率。
所有制剂都导致所有测试的细胞系增殖的浓度依赖性抑制(图15)。DTX制剂比PTX制剂的细胞毒性更大,尽管组间没有显著性差异(P>0.05,单因素方差分析)。的IC50比的IC50低约2-5 倍。发现负载PTX与DTX的HPG-C8/10-MePEG纳米颗粒分别与商品化制剂与的细胞毒性一样(图15)。对照的HPG-C8/10-MePEG纳米颗粒(无药物)显示在15-1,500nM的浓度范围内无细胞毒性(数据未显示),而与Tween 80已显示了即使在低浓度下对细胞的毒性(Iwase K,Oyama Y,Tatsuishi T等,ToxicolLett,154(1-2),143-148(2004);Henni-Silhadi W,Deyme M,Boissonnade MM等,Pharm Res,24(12),2317-2326(2007))。
实施例13:膀胱内的PTX与DTX制剂在膀胱癌原位模型中的功效
在总数为60只裸鼠中完成体内研究以评估膀胱内的(1mg/ml,Bristol-Myers-Squibb);(0.5mg/ml,Sanofi-Aventis);负载PTX(1mg/ml)的HPG-C8/10-MePEG;以及负载DTX(0.5mg/ml)的HPG-C8/10-MePEG在膀胱癌的小鼠异种移植模型中的功效。已报道过这一原位的小鼠模型(Mugabe C,Hadaschik BA,Kainthan RK等.BJU Int,103(7),978-986(2009);Hadaschik BA,Black PC,Sea JC等.BJU Int,100(6),1377-1384(2007);Hadaschik BA,ter Borg MG,Jackson J等.BJU Int,101(11),1347-1355(2008))。根据加拿大动物管理委员会要求进行动物研究。用异氟烷麻醉11周大小的雌性裸鼠(Harlan,Indianapolis,IN)。在将润滑的24号Jelco血管导管(Medex Medical Ltd.,Lancashire,UK)穿过尿道至膀胱之前将表浅的6-0聚丙烯荷包缝合置于尿道口周围。膀胱的PBS单一冲洗后,灌注200,0000个KU7-荧光素酶细胞作为50μl单一细胞悬浮液并且将荷包缝合结扎2.5小时。为定量体内肿瘤负载,在4,11,18,25以及33天腹膜内注射150mg/kg荧光素后,用IVIS200成像系统(Xenogen/Caliper Life Sciences,Hopkinton,MA)在仰卧位对动物成像15分钟。利用Living Image软件(Xenogen)获取及分析数据。在肿瘤接种后第5天,小鼠随机接受50μlPBS(对照);HPG-C8/10-MePEG(无药物);(1mg/ml);负载PTX(1mg/ml)的HPG-C8/10-MePEG;0.5mg/ml);负载DTX(0.5mg/ml)的HPG-C8/10-MePEG膀胱内处理。在肿瘤接种后第5 天和第19天,给予膀胱内治疗。生物荧光水平在组中是相等的,然而,因为肿瘤因个体的小鼠而异,为统计分析,每只小鼠中在第4天,以最初的通量做参比,将处理后的肿瘤生物荧光标准化。肿瘤接种后第33天进行尸检。移除整个膀胱,固定于10%缓冲的福尔马林并且埋入石蜡中。制备5μm切片并且利用标准的技术用H&E染色。在BLISS显微镜成像工作站(Bacus Laboratories Inc.,Lombard,IL)上评估及扫描所有的载玻片。KU7-荧光素酶癌细胞膀胱内接种后,所有的小鼠发展为膀胱肿瘤。然而,两只小鼠没有从麻醉中恢复过来并且在死于肿瘤接种的同一天。发现一只组的小鼠在成像的最后一天死亡(肿瘤接种后第33天),在本组早先的测量中这只小鼠具有最大的肿瘤。由于不可逆的体重减轻(15%体重减轻),对在负载DTX的HPG-C8/10-MePEG中另一只小鼠实行安乐死。为统计分析,将这些小鼠排除在研究之外。总的来说,小鼠都很好地耐受了膀胱内的PTX与DTX,无论商品化与还是HPG-C8/10-MePEG制剂,并且没有观察到主要的毒性或体重减轻。在肿瘤接种后第5天及第19天给予膀胱内治疗。与对照组小鼠(PBS&空白的HPG-C8/10-MePEG)相比,负载PTX与DTX的HPG-C8/10-MePEG显著地抑制了肿瘤生长(P<0.001,双因素方差分析,Bonferroni测试后)(图16)。在与对照组比较时,不同于(1mg/ml)显著地减少了肿瘤生长(P<0.01,双因素方差分析,Bonferroni测试后)。然而在与 处理组间未观察到显著的差异。基于早先关于PTX的报道(Mugabe C,Hadaschik BA,Kainthan RK等.BJU Int,103(7),978-986(2009))选择PTX(1mg/ml)与DTX(0.5mg/ml)的膀胱内灌注剂量并且体外细胞毒性数据显示DTX比PTX更有效(图15)。研究的最后,负载PTX与DTX的HPG-C8/10-MePEG纳米颗粒中的肿瘤生长分别被抑制了87%与97%,与PBS对照组相比。与分别显示了43%与65%肿瘤生长抑制。每一处理组中小鼠随时间变化的代表性生物荧光成像显示于图17中。膀胱组织的组织学检查显示KU7-荧光素酶肿瘤展现了积极的生长模式与频繁的多灶性,但是肿瘤接种后33天,处理组中大多数的肿瘤通常限于固有层及与高级pT1阶段疾病有 关(图18)。
推测HPG的低纳米大小范围可允许粘蛋白链间渗透并且与尿路上皮的雨伞细胞接触,导致这些纳米颗粒至包括肿瘤组织的膀胱壁增强的胞吞作用。也可能的是HPG上表面的MePEG链可通过链缠结与粘蛋白糖蛋白接触,导致这些纳米颗粒陷入粘蛋白层中,引起负载药物的纳米颗粒在膀胱中延长的停留时间。表7显示负载PTX与DTX的HPG-C8/10-MePEG纳米颗粒展现了比商品化制剂或高2-3倍的膀胱组织积聚。
实施例14:药代动力学
为评估膀胱内PTX与DTX制剂的药代动力学特性,用(1mg/ml,n=3);(0.5mg/ml,n=4);负载PTX的HPG-C8/10-MePEG(1mg/ml,n=4);和/或负载DTX的HPG-C8/10-MePEG(0.5mg/ml,n=4)灌注小鼠。在膀胱内灌注后0、30以及60分钟时取尾部血液样品。2小时后,利用CO2窒息杀死所有的小鼠并且通过心脏穿刺除去额外的血液。在微血细胞比容管(Fisher Scientific,Pittsburg,PA)或血清分离管(Becton Dicknson)中离心血液样品并且在液氮中快速冷冻血清。也收获每只小鼠的尿液和膀胱并且在冷冻前,将膀胱切开以暴露内腔,并且在五次连续的10ml PBS洗涤液中强力地洗涤。所有样品储存于-80℃。由集成的超高压液相色谱(Waters Acquity UPLC)分离系统组成的UPLC-MS/MS系统用于分析,结合利用Waters TQD质谱仪的质谱分析。在以下情况下操作系统:电喷射离子源阻断温度150℃,脱溶剂温度350℃,采样锥电压45kV,毛细管电压0.70kV,提取器电压3kV,RF电压0.1kV,进样锥气体流速25l/h,脱溶剂气体流速600l/h以及碰撞气体流速0.2ml/min。分子经受阳离子模式的电喷雾离子阱。通过溶剂/溶剂提取方法从小鼠血清提取DTX。将50μl等分的小鼠血浆和标准液与150μl含0.1%甲酸的乙腈混合于96孔平板并且在室温下涡旋1分钟。在4℃下以5,500转速离心样品10分钟。然后将100μl上清液与50μl蒸馏水混合,混合并且涡旋30秒。称重膀胱组织并且在0.1%甲酸/甲醇中利用氧化锆珠(Biospec Products)及装备有微量试管架的微型珠搅拌器(Biospec Products)均质化60秒。在4℃下以14,000转速(AllegraTM 25R centrifuge,Beckman-Coulter)离心样品2分钟。添加150μl含0.1%三氟乙酸的甲醇至样品,混合并且在4℃下以14,000转速(AllegraTM25R centrifuge,Beckman-Coulter)涡旋15分钟。利用UPLC-MS/MS进行所有的样品分析。DTX的定量限为10ng/ml,具有从加入标准的对照样品97%的恢复。
几个血清样品具有不可计量的或不可检测的水平。通常,PTX与DTX的血清水平是低的(5-20ng/ml),随后膀胱内灌注。血清水平在不同组间和/或在不同的时间点没有显著的差异(P>0.05)(表7)。然而,膀胱组织水平比血清水平高约100-500倍。负载PTX与DTX的HPG-C8/10-MePEG纳米颗粒显示了比商品化制剂高2-3倍的膀胱组织积聚,尽管差异没有统计学意(P>0.05,单因素方差分析,Bonferroni’s多重比较检定)。尿液中最终的药物浓度比初始的计量溶液低约3-5倍。这归因于在膀胱内灌注2小时期间尿液稀释。然而,PTX与DTX的最终尿液浓度在不同的处理组间没有显著的差异(P>0.05,单因素方差分析)。通常,PTX与DTX的血清水平是低的(5-20ng/ml)随后膀胱内灌注。血清水平在不同组间和/或在不同的时间点没有显著的差异(P>0.05)(表7)。
表7膀胱内PTX与DTX制剂在原位异种移植中的药代动力学
1膀胱内灌注2小时后通过HPLC所测量的小鼠尿液中最终浓度。
22小时膀胱内灌注后通过LC/MS/MS所测量的小鼠膀胱组织中的PTX或DTX浓度。
3取自膀胱内灌注后1小时及2小时通过LC/MS/MS所测量的小鼠血清中PTX或DTX的浓度。
所示数据为均值±标准差(SD)。
实施例15:HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2的合成
根据实施例7中所述的方案制备HPG-C8/10-MePEG。1H NMR(400MHz,D6-DMSO)δH:0.75-0.82(-CH3,TMP);0.82-0.92(-CH3-烷基在O/DGE上);1.15-1.55(-CH2-,烷基在O/DGE上);2.50(溶剂,D6-DMSO);3.15-3.80(-CH与-CH2-,来自HPG内核);3.23(-OCH3-来自MePEG),3.32(残留水);4.8(-OH)。利用下列程序合成具有胺基取代的各种靶标量的HPG-C8/10-MePEG-NH2。所用试剂的量总结于表8中。靶标胺基取代代表NH2摩尔的靶标号/HPG摩尔并且通过HPG-C8/10-MePEG-NH2(X)表示,其中x为NH2的61,121及161摩尔/HPG摩尔。HPG-C8/10-MePEG溶解于15ml无水1,4-二氧己环。用无水己烷漂洗氢化钾(KH)三次以除去矿物油并且在真空下干燥。聚合物溶液与KH组合并且在室温下搅拌直至形成澄清的溶液。通过溶解于二氯甲烷并且在Na2SO4或MgSO4上搅拌过夜干燥N-(2,3-环氧丙基)-苯邻二甲酰亚胺)(EPP)(Sigma-Aldrich)。过滤溶液并且在真空下干燥以除去二氯甲烷。将干燥的EPP溶解于无水1,4-二氧己环并且添加至聚合物在85-90℃下搅拌过夜。通过将其穿过阳离子交换树脂(Amberlite IRC-150)三次中和产物,然后从乙醚沉淀三次以除去未反应的EPP。通过与水合肼的肼解作用实现苯邻二甲酰亚胺功能的剪切。添加过量的水合肼溶液(2ml)至含聚合物的甲醇溶液中并且回流混合物72小时。回流后,蒸发甲醇,利用10,000MWCO膜,水作参比,透析聚合物48小时并且冷冻干燥。1HNMR(400MHz,D6-DMSO)δH:0.75-0.82(-CH3,TMP);0.82-0.92(O/DGE上-CH3-烷基);1.15-1.55(-CH2-,O/DGE上烷基);2.50(溶剂,D6-DMSO);2.60-2.80(-CH与-CH2-,来自HPG内核);3.15-3.80(-CH与-CH2-,来自HPG内核);3.23(-OCH3-来自 MePEG)。用于具有N-(2,3-环氧丙基)-苯邻二甲酰亚胺(EPP)的HPG-C8/10-MePEG聚合物上一些羟基(10-20%)的表面修饰,随后通过肼解作用剪切苯邻二甲酰亚胺功能基团以产生HPG-C8/10-MePEG-NH2的反应方案总结于Scheme VII(R,代表基于烷基(C8/C10)链混合物的疏水性内核。()7,代表基于MePEG 350的亲水性外壳)。
选择HPG-C8/10-MePEG-NH2(121)用于药物负载及进一步的动物研究中的评估。
NMR与GPC
自利用氘化溶剂的Bruker Avance 400MHz(磁场强度9.4T)NMR光谱仪(Cambridge Isotope Laboratories,99.8%D)所记录的异核单量子相关谱(HSQC)NMR实验估算MePEG与HPG上烷基链的分数。通过具有多角度激光散射检测的凝胶渗透色谱法(GPC-MALLS)确定聚合物的分子量与多分散性。
图19A显示了HPG-C8/10-MePEG的代表性2维HSQC光谱。通过2维HSQC光谱确认具有N-(2,3-环氧丙基)-苯邻二甲酰亚胺)HPG-C8/10-MePEG的表面修饰,其中鉴定芳族的苯邻二甲酰亚胺CH基团(1H化学位移7.2-7.8ppm&13C化学位移125-135ppm)。通过1维NMR与2维HSQC实验监控通过肼解作用剪切苯邻二甲酰亚胺基团以产生游离胺基的成功。图19B显示了HPG-C8/10-MePEG-NH2(121)的代表性2维HSQC光谱,其中显示成功地剪切苯邻二甲酰亚胺保护基团以产生胺基(2.60-2.80ppm&13C化学位移45ppm,图19B)。HSQC NMR数据确认dHPG的结构为具有光谱中明显的分支体系结构的超支化聚合物(图19)。可从HSQC实验中的信号积分计算C8/10烷基链与dHPG上MePEG的摩尔分数。通过比较MePEG甲氧基和辛基/癸基缩水甘油醚(O/DGE)甲基的积分与TMP CH3基团的积分,计算对每个dHPG聚合物而言的MePEG分数以及O/DGE(mol/mol)(表10)。
电导滴定和荧光胺测定
通过利用HCl和NaOH的电导滴定测量HPG-C8/10-MePEG聚合物上衍生的胺基的摩尔分数。25℃下在YSI模型35电导度计及具有铂电极的3403电池上完成电导滴定。注射泵(Harvard Instruments)用于以恒定的流量0.0102ml/min注射稀释的NaOH溶液。对典型的滴定而言,将大约10mg HPG-C8/10-MePEG-NH2溶解于蒸馏水中并且首先用0.05N HCl滴定,随后用0.05N NaOH回滴定。每隔30秒测量溶液的电导。邻苯二甲酸氢钾(0.05N)用于标准化氢氧化钠溶液。基于电导滴定与分子量测量,计算胺基/HPG分子摩尔数量并且所获得的值在50-119mol/mol的范围,所述值由NH2/HPG-C8/10-MePEG-NH2摩尔的靶向摩尔比率组成(表8&10)。
表8用于HPG-C8/10-MePEG-NH2合成的试剂的化学计量学
KH,氢化钾;EPP,N-(2,3-环氧丙基)-苯邻二甲酰亚胺。HPG-C8/10-MePEG的分子量特性,Mw=83,000并且Mw/Mn=1.22。
也可利用荧光及分子量测量计算胺基/HPG分子的摩尔数量。例如,可使用用于NH2的荧光胺测定(表9)。通过测量>5mg HPG-NH2至2ml LC/MS玻璃小瓶并且添加适当量的去离子水以产生浓缩的储备溶液,然后对混合物进行声处理直至将HPG-NH2溶解,将储备溶液稀释至1mg/ml去离子水/HPG-NH2mg,制备HPG-NH2样品。
通过测量大约1mg苯丙氨酸至铝微重盘并且将其转移至20ml玻璃小瓶,然后添加5ml去离子水/苯丙氨酸mg,对混合物进行声处理直至将苯丙氨酸溶解(0.2mg/mL,苯丙氨酸储备溶液)制备苯丙氨酸标准液。将60μL0.2mg/ml苯丙氨酸储备溶液转移至玻璃的LC/MS小瓶。添加90μL去离子水,并且涡旋以混合溶液(80ng/mL苯丙氨酸起效标准液)。
将40μL 1mg/mL HPG-NH2储备液转移至96孔平板,添加10μL去离子水(或添加50μL,40μL,30μL,20μL,10μL或0μL 80ng/mL苯丙氨酸起效标准液并且用去离子水加满至50μL,如果有必要的话);用移液器吸取样品混合。添加12.5μL硼酸钠缓冲液至平板并且用移液器吸取混合。吸管端用0.03%荧光胺溶液漂洗两次以包被尖端及防止滴水。添加12.5μL 0.03%荧光胺溶液至样品孔。用移液器吸取样品混合然后将其放在平板振动器上,覆盖并摇晃1分钟。添加175μL去离子水以与过量的荧光胺反应并且用移液器吸取混合,并且暂时地置于平板振动器上。通过荧光平板读取器,设定390nm激发波长,475nm发射波长以及激发与发射波长二者的5nm带宽,分析样品。
表9通过荧光胺测定所测量的胺摩尔/HPG摩尔
aEPP-A(EPP来自于Atlantic)纯度:通过UPLC,78%以及通过NMR 69%。
bEPP-S(EPP来自于Sigma-Aldrich)纯度:通过UPLC,91%以及通过NMR 95%。
热分析
差示扫描量热法(DSC)与热重量分析法(TGA)用于评估dHPG的热及降解特性。利用TA Instruments DSC Q100及TGA Q50进行热分析。通过使称重的样品在气密封口的铝锅上以10℃/分钟,-90℃-85℃温度范围内通过“热-冷-热”循环获得DSC游程数。TGA游程数主要以“逐步等温的”模式进行,其中在等温条件下观察降解过程中每一重量减轻相并且将HPG加热直到500℃下接近100%重量减轻。表10显示了HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2样品的热事件。HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2样品显示了相似的玻璃化DSC/TGA属性。HPG显示玻璃化转变温度在-45℃--58℃。胺基的存在产生了HPG的Tg有小幅上升。在300℃以上温度观察主要的降解事件,其显示了HPG良好的热稳定性特性。大约3-5%重量减轻发生在温度低于100℃,很可能是由于HPG中一些残余的溶剂或水。HPG良好的热稳定性适合于药物应用以允许热灭菌方法的潜在用途。
颗粒大小与Z电势
利用Malvern NanoZS颗粒大小分析仪进行颗粒大小与Z电势分析,每一分析使用DTS0012一次性定径比色皿。用0.2μm直列式注射器式滤器过滤样品(PALL Acrodisc具有13mm尼龙膜)。样品采集参数为:角度为具有自动衰减的173°反散射;游程数11(10s/游程);分散剂为25℃水(粘性0.8872cP,阻力指数(RI)1.330);Mark-Houwink参数A=0.428及K=7.67e-05(cm2/s)。假定dHPG具有与RI=1.460及0.01 吸收的聚乙二醇(PEG)相似的折射率。最终的数据代表所有游程的平均值。HPG为具有流体力学半径<10nm的小纳米颗粒。HPG形成小于10nm极其小的纳米颗粒。表10显示了HPG的颗粒大小及Z电势特征。具有胺基的HPG-C8/10-MePEG的表面衍生对它们的颗粒大小没有影响,然而,观察到对Z电势有显著的影响。端胺基HPG聚合物的Z电势在低pH时高度正性,并且在碱性状态中转变为轻微的负性值。表面电荷中这一pH可确定的改变由胺基的质子化/去质子化引起。在生理的pH7.4时,电离了HPG上一些胺基,并且因此,预期正性的Z电势(表10)。然而,在pH值大于8时基本上所有的胺基都是不带电荷的从而在pH11时所观察到的轻微的负电荷很可能是由于这些HPG上存在的带负电的羟基。HPG的药物负载对它们的颗粒大小没有显著的影响并且HPG仍然作为单分子的胶束良好地分散于溶液中。负载DTX的HPG纳米颗粒为物理性质稳定的,在室温下1周储存期间没有观察药物沉淀或聚集。
表10用C8/10烷基链衍生以及用MePEG与胺基修饰的HPG的物理化学特征
*选择这一批用于药物负载及体内研究;1Tg,取自转变中点的玻 璃化转变;2Td,取自最大的重量减轻时降解温度。
实施例16:粘膜粘附特性的评估
为评估HPG的粘膜粘附特性,使用Thongborisute与Takeuchi研发的粘蛋白颗粒方法(Thongborisute,J.;Takeuchi,H.Int.J.Pharm.2008,354,204-209)。该方法基于颗粒大小的改变,所述颗粒大小的改变由于作为粘附的聚合物与粘蛋白间相互作用结果的超微粘蛋白聚集。将超微粘蛋白溶液与相同体积的HPG(10%w/v)在100mM醋酸缓冲液中混合,在37℃下涡旋及孵育30分钟。通过利用3000HS Zetasizer(Malvern Instruments,San Bernardino,CA)的光散射测量监控颗粒大小的改变。每一测试以一式三份的进行并且将壳聚糖溶液(1%w/v)用作阳性对照。在与壳聚糖(1%w/v)或HPG-C8/10-MePEG-NH2(121)(10%w/v)溶液孵育后,粘蛋白的颗粒大小显著地增加(图20)。HPG-C8/10-MePEG(10%w/v)对粘蛋白颗粒的大小没有影响。超微粘蛋白增加的颗粒大小是由于聚集的粘蛋白颗粒与HPG-C8/10-MePEG-NH2纳米颗粒并且可归因于粘蛋白与胺取代的HPG间的粘膜粘附力。壳聚糖,众所周知的粘膜粘附的聚合物,被用作阳性对照。然而,由于它的高分子量及水溶液中低溶解度,使用壳聚糖的稀释溶液(1%w/v)。甚至共孵育后,该稀释溶液在粘蛋白的颗粒大小中表现出显著的改变。据信壳聚糖与HPG-C8/10-MePEG-NH2的粘膜粘附是由于正电荷的胺基与负电荷的粘蛋白颗粒间静电相互作用,而且其他的因素诸如氢键结合,疏水效应以及链缠结也有影响。然而,HPG-C8/10-MePEG的粘膜粘附的缺乏提示静电吸引好像是HPG-C8/10-MePEG-NH2的粘膜粘附特性的主要因素。
实施例17:细胞增殖/结合以及摄取研究
细胞增殖
KU7-荧光素酶以5,000个细胞/孔沉积于96孔平板100μl体积的补充了10%FBS的McCoy’s介质中并且允许在添加新鲜制备的HPG-C8/10-MePEG和/或HPG-C8/10-MePEG-NH2(溶解于PBS,pH 7.4, 0-150μg/ml)溶液前,平衡24小时。细胞暴露于HPG溶液2小时并且利用早先所述的CellTiter96水非放射性细胞增殖测定(Promega,Madison,WI)(Mugabe,C.;Hadaschik,B.A.;Kainthan,R.K.;Brooks,D.E.;So,A.I.;Gleave,M.E.;Burt,H.M.BJU Int.2009,103,978-986),72小时后确定细胞活力。
HPG的若丹明标记
用如早先报道的(Savic,R.;Luo,L.;Eisenberg,A.;Maysinger,D.Science 2003,300,615-618)四甲基若丹明-羰基叠氮化物(TMRCA)共价地标记HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2(121)聚合物。四甲基若丹明-羰基叠氮化物(TMRCA)购于Invitrogen Canada Inc.(Burlington,ON)。简而言之,将500mg HPG(HPG-C8/10-MePEG或HPG-C8/10-MePEG-NH2(121))溶解于5ml无水的1,4-二氧己环。将适当量的四甲基若丹明-5-羰基叠氮化物(TMRCA,MW 455.47)溶解于2ml无水的1,4-二氧己环以产生最终的浓度1mg/ml。将该675μl荧光探针的等分,其对应大约20mol%HPG,添加至HPG溶液并且在80℃下,氮气流下,油浴中加热并且搅拌5小时。通过DMF(MWCO12,000-14,000)作参比,透析直至透析液为无色的,然后蒸馏水作参比,透析24小时,除去未反应的探针。冷冻干燥荧光标记的聚合物(HPG-TMRCA)并且储存于-80℃下琥珀色小瓶中。
细胞结合与摄取
KU7-荧光素酶细胞用于评估若丹明标记的HPG的结合及摄取。细胞以10,000个细胞/孔沉积于96孔平板100μl体积的补充了10%FBS的McCoy’s介质中并且允许平衡24小时。除去介质并将细胞与若丹明标记的HPG(HPG-C8/10-MePEG-TMRCA或HPG-C8/10-MePEG-NH2(121)-TMRCA,1.56-200μg/ml)孵育2小时。孵育期后,用PBS洗涤细胞3次并且用200μl 0.5%Triton X-100(PBS中pH 8)溶解细胞而且通过荧光光谱(Synergy 4.0)以激发/发射545/578测量细胞的荧光结合量。标准液制备于Triton X-100与PBS(pH 8)中若丹明标记的HPG(0.781-6.25μg/ml)。细胞摄取或表面结合的若丹明标记的HPG量表示为所添加至每一孔上聚合物的总量的百分比。
细胞摄取的共聚焦荧光分析
KU7-荧光素酶细胞生长于10cm培养皿,盘底上1cm x 1cm盖玻片用于细胞在其上生长,直至达到~75%聚焦,其对应于7x 104个细胞的细胞量。然后除去包含细胞的盖玻片并且用温暖的PBS洗涤3次。然后在整个摄取测定的时期,放置盖玻片在石蜡衬里的培养皿中细胞侧朝上。添加若丹明标记的HPG聚合物(250μl HPG-C8/10-MePEG-TMRCA或HPG-C8/10-MePEG-NH2(121)-TMRCA)至盖玻片上浓度为1mg/ml的细胞。孵育细胞1,4,8以及24小时。在每一时间点后,PBS中强力地洗涤盖玻片4次。在轻轻地吸去过量的PBS后,室温下将250μl 3.7%多聚甲醛用于固定细胞10分钟。然后在PBS中洗涤盖玻片3次以上,沉入水中,吸去过量的液体,并且最终用4',6-二脒基-2-苯基吲哚(DAPI)及Prolong gold在载玻片上固定细胞侧朝下。将清澈的指甲油少量地用于盖玻片边缘周围以停止样品干燥。过夜孵育确保样品的适当硬化,然后其为成像作好准备。在Olympus FV-1000反相共聚焦显微镜上进行显微研究。所用的激光波长为568nm与405nm,分别用于若丹明与DAPI的成像。也进行直接对比(DIC)以显现细胞膜并且同样地用405nm激光激活。保持每一聚合物组中激光功率与高电压增益相对地恒定以允许一致的比较。为了清楚地显示标记的聚合物在细胞内部,通过荧光与DIC分析图像。
细胞增殖/结合及摄取研究
低浓度时,广泛地结合HPG-C8/10-MePEG-NH2(121)-TMRCA纳米颗粒并且内化至KU7-荧光素酶,然而在HPG-C8/10-MePEG-TMRCA纳米颗粒浓度低于12.5μg/ml时没有观察到细胞结合或摄取的迹象(图21A)。该聚合物的强结合属性很可能由于带正电荷的HPG-C8/10-MePEG-NH2(121)聚合物与带负电荷的KU7-荧光素酶的细胞膜间的静电吸引。然而,随着HPG浓度的增加,HPG-C8/10-MePEG-NH2(121)-TMRCA纳米颗粒的细胞结合与摄取达到了饱和点(处于25μg/ml)而发现HPG-C8/10-MePEG-TMRCA纳米颗粒至KU7-荧光素酶的细胞结合与摄取在浓度12.5μg/ml-50μg/ml时观察到浓度依赖性的线性关系,随后在较高的聚合物浓度时没有那么明显 的结合与摄取(图21A)。为评估HPG-C8/10-MePEG-NH2(121)-TMRCA纳米颗粒的饱和行为是否由于它们的细胞毒性效应,我们评估HPG对KU7-荧光素酶细胞增殖的影响。将细胞暴露于空白的(无负载药物)HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2纳米颗粒(0-150μg/ml)2小时并且通过MTS测定确定细胞活力。HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2纳米颗粒显示了相似的增值效应并且在测试浓度下与KU7-荧光素酶细胞系为生物相容性的(图21B)。因此,所观察到的HPG-C8/10-MePEG-TMRCA与HPG-C8/10-MePEG-NH2(121)-TMRCA纳米颗粒在细胞结合及摄取中的差异很可能不是由于它们对KU7-荧光素酶细胞系的细胞毒性效应。
若丹明标记的HPG的内化的共聚焦荧光分析
共聚焦显微镜用于监控纳米颗粒是否被细胞内化或简单地结合KU7-荧光素酶的细胞膜。HPG-C8/10-MePEG-TMRCA与HPG-C8/10-MePEG-NH2(121)-TMRCA纳米颗粒迅速地被KU7-荧光素酶细胞内化并且通过1小时的孵育获得完全的摄取(数据未显示)。通过荧光分析在细胞质中观察到若丹明标记的HPG的存在。与仅仅粘附或存在于细胞膜相反,观察到HPG-C8/10-MePEG-TMRCA和/或HPG-C8/10-MePEG-NH2(121)-TMRCA纳米颗粒遍及细胞质的存在。没有在KU7-荧光素酶细胞的细胞核间中检测到来自聚合物的荧光。HPG纳米颗粒在细胞核间中的缺乏可能由于它们的相对高的分子量(<80kDa)。当与对照的细胞在所有的时间点比较时,HPG-C8/10-MePEG-TMRCA与HPG-C8/10-MePEG-NH2(121)-TMRCA纳米颗粒对KU7-荧光素酶细胞的活力及普及没有影响。总之,KU7-荧光素酶细胞至1小时的孵育期摄取若丹明标记的HPG纳米颗粒并且在1,4,8或24小时时间点所获得的图像中没有差异。
实施例18:HPG中DTX的负载与定量以及HPG释放的DTX
HPG中DTX的负载与定量
通过溶剂蒸发方法,其中药物与聚合物溶解于普通的有机溶剂中并且除去溶剂,HPG-C8/10-MePEG与HPG-C8/10-MePEGNH2(121)负载DTX。由此得到的聚合物/药物基质与10mM PBS(pH7.4)重组。由此得到的溶液通常为澄清的但是如果观察到白色的颗粒,将溶液离心(18,000g,10min)并且将上清液转移至新的小瓶。
通过反相HPLC确定DTX掺入HPG的量。利用对称的C18柱(Waters Nova-Pak,Milford,MA)与含乙腈、水及甲醇(58:37:5,v/v/v)混合物的1ml/min流速的流动相进行药物含量分析。样品注射体积为20μl并且利用波长232nm的UV检测进行检测。总运行时间设定为5分钟并且DTX保留时间为2.9分钟。通过该方法获得多达5%w/w药物负载,其中对应约5-6个DTX分子/HPG分子。DTX的水溶解性在7μg/ml的范围内(Du,W.;Hong,L.;Yao,T.;Yang,X.;He,Q.;Yang,B.;Hu,Y.Bioorg.Med.Chem.2007,15,6323-6330;Liggins,R.T.;Hunter,W.L.;Burt,H.M.J.Pharm.Sci.1997,86,1458-1463)并且HPG中掺入DTX导致该药物的水溶解性增加大约1000倍。
HPG释放的DTX
通过透析方法确定HPG纳米颗粒(HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2(121))释放的DTX。简而言之,称量100mg HPG-C8/10-MePEG或HPG-C8/10-MePEG-NH2(121)并且将其与DTX混合在1ml乙腈溶液中,加入标准的15μl Ci 3H-DTX,然后在氮气流下干燥以除去溶剂。HPG/DTX基质与2ml PBS形成水合物并且将其转移至透析袋,以500ml人造尿液(pH6.5)作参比,透析及以100转速摇动。利用0.1M HCl将溶液的pH调至6.5。选择pH6.5,因为它是人尿液生理学范围的中间值,尽管它可能在广泛的范围内(pH4.5-8)变化(Brooks,T.;Keevil,C.W.Lett.Appl.Microbiol.1997,24,203-206)。在不同的时间点,测量透析袋的体积并且取出10μl样品用于透析袋中残余的放射性的测量。将整个的外部释放介质与新鲜的介质交换以维持下沉状态。通过β-闪烁计数(Beckman Coulter Canada,Mississagua,ON)确定透析袋中残余的3H-DTX在每一时间点上的浓度。通过从实验开始时最初的药物量减去每一时间点上残余的药物量计算所释放的药物累积百分比。数据表示为根据时间所释放的药物累积百分比。通过具有很少或没有释放的突发相的连续控制的释放表征来自HPG的DTX 的释放属性(图22)。在孵育的第一个24小时内释放大约55%最初密封的药物。HPG表面上胺基的存在对药物释放没有影响(图22)。
实施例19:评估原位膀胱癌模型中的膀胱内DTX制剂
评估膀胱内负载DTX的HPG制剂在具有原位膀胱异种移植的小鼠中的耐受性和功效。已报道了所用的原位小鼠模型(Mugabe,C.;Hadaschik,B.A.;Kainthan,R.K.;Brooks,D.E.;So,A.I.;Gleave,M.E.;Burt,H.M.BJU Int.2009,103,978-986;Hadaschik,B.A.;Black,P.C.;Sea,J.C.;Metwalli,A.R.;Fazli,L.;Dinney,C.P.;Gleave,M.E.;So,A.I.BJU Int 2007,100,1377-1384;Hadaschik,B.A.;ter Borg,M.G.;Jackson,J.;Sowery,R.D.;So,A.I.;Burt,H.M.;Gleave,M.E.BJU Int.2008,101,1347-1355)。根据加拿大动物管理委员会的要求进行所有的动物研究并且来自我们机构的动物管理协议已得到了动物管理委员会的批准(英属哥伦比亚大学)(The University of British Columbia)。在该模型中,使用表达KU7-荧光素酶癌细胞的荧光素酶。为进行肿瘤接种,用异氟烷麻醉8周大小的雌性裸鼠(Harlan,Indianapolis,IN)。在润滑的24GJelco血管探针(Medex Medical Ltd.,Lancashire,UK)穿过尿道至膀胱之前将表浅的6-0聚丙烯荷包缝合置于尿道口周围。膀胱的PBS单一冲洗后,灌注200,0000个KU7-荧光素酶细胞作为50μl单一细胞悬浮液并且用荷包缝合结扎2.5小时,在此期间保持小鼠处于麻醉状态。缝合除去后,将小鼠置于笼中并且监控至它们恢复知觉并且以正常的方式排泄。肿瘤接种后5天,经由膀胱内灌注(50μl,2h停延时间),根据下列的处理组:PBS(对照);(0.5与1.0mg/ml,含DTX的Tween 80);含DTX的HPG-C8/10-MePEG(0.5与1.0mg/ml);含DTX的HPG-C8/10-MePEG-NH2(121)(1.0mg/ml)处理26只随机选择的小鼠。在处理的第一天并且其后每天监控小鼠几个小时。报道毒性的任何迹象,尤其是重量减轻,摄食饮水的改变,昏睡,弓背姿势,和/或压力的总体表现。牺牲24小时内没有恢复过来的显示疼痛或疾病迹象的任一小鼠。通过用IVIS 200成像系统(Xenogen Corp.,Alameda,CA)在2,8,12以及19天小鼠的非侵润性成像监控肿瘤负担。简而言之,腹膜内注射 小鼠150mg/kg荧光素,用异氟烷麻醉并且在荧光素注射后恰好15分钟时仰卧位成像。利用Living Image软件版本2.50(Xenogen)获得数据。
肿瘤接种后2天,所有的小鼠发展为膀胱肿瘤,然而也有2只小鼠发展为如生物荧光成像所示的肾脏肿瘤(图23A)。总之,小鼠很好地耐受了膀胱内DTX,无论是商品化还是HPG制剂。没有观察到主要的毒性并且所有小鼠存活至研究期末。然而,肿瘤接种后第8天,一些小鼠体重减轻了约5%,尽管它们在下一周恢复了。体重减轻可能为膀胱内处理和/或处理后每天食物与水摄取较少的结果。然而,不同组间的体重减轻没有显著的差异(p>0.05)。
选择0.5mg/ml与1.0mg/ml的剂量以建立膀胱内DTX在具有膀胱癌异种移植的小鼠中适当的给药方案。单一剂量处理的小鼠,无论是1.0mg/ml的0.5mg/ml含DTX的HPG-C8/10-MePEG,还是1mg/ml的HPG-C8/10-MePEG和/或HPG-C8/10-MePEG-NH2(121)都强有力地抑制了肿瘤生长。在肿瘤接种后第19天,所有处理组,除了 (0.5mg/ml)之外,都显示了与PBS对照比较有统计学意义的肿瘤抑制(p<0.001,post-hoc Bonferoni analysis双因素方差分析后)。
据信这些纳米颗粒的粘膜粘附特性增加了与尿路上皮接触的亲密性,导致了增强的药物渗透性并且摄取至膀胱壁,这可能由于尿路上皮的紧密连接或脱皮的调整。由于它们极小的尺寸(Rh<10nm),HPG可扩散穿过粘蛋白糖蛋白并且与尿路上皮的雨伞细胞直接相互作用,导致了膀胱壁或肿瘤组织对这些纳米颗粒增强的胞吞作用。
进行第二次研究以评估较低灌注剂量的HPG中DTX的有效性。为进行该研究,使用没有明显的膀胱肿瘤或具有低水平的生物荧光的小鼠,如通过IVIS 200成像系统在第19天所确定的。总计发现12只小鼠适合于如上文所述的第二次肿瘤再接种。与早先研究中的100%比较,肿瘤占据约75%并且可归因于免疫反应,因为早先用相同的细胞系接种小鼠;尽管使用无胸腺的免疫功能不全的小鼠,这些小鼠仍然具有固有的巨噬细胞和自然杀伤细胞所表征的局部免疫系统。来自于再接种后发展为膀胱肿瘤的9只小鼠,2只小鼠甚至发展为更大的(10-100倍)膀胱肿瘤。肿瘤再接种后第5天,随机分配发展为膀胱肿 瘤的小鼠成2组以接受单一的50μl膀胱内负载DTX(0.2mg/ml)的HPG-C8/10-MePEG(n=5)或HPG-C8/10-MePEG-NH2(121)(n=4)。肿瘤再接种后5天、11天及19天将小鼠成像。膀胱内负载DTX的HPG-C8/10-MePEG-NH2(121)抑制小鼠中的肿瘤生长而负载DTX的HPG-C8/10-MePEG在相同浓度下不能实现如此情形。肿瘤再接种后第11天和第19天,4只小鼠中的3只没有表现出肿瘤生长的迹象,随后用负载DTX(0.2mg/ml)的HPG-C8/10-MePEG-NH2(121)纳米颗粒单一的膀胱内处理而用负载DTX(0.2mg/ml)的HPG-C8/10-MePEG纳米颗粒处理的5只小鼠中的4只显示出膀胱肿瘤生长的迹象并且1只小鼠还发展成为肾脏肿瘤(图24)。再次,小鼠很好地耐受了这些制剂,在这些研究期间没有发生主要的毒性或体重减轻。
实施例20:体外细胞毒性研究
评估商品化制剂与负载DTX的HPG制剂抗KU7-荧光素酶细胞系、低级的(RT4,MGHU3)与高级的(UMUC3)人尿路上皮癌细胞系的细胞毒性效应。
(含DTX的Tween 80)购于Sanofi-Aventis Canada Inc.(Laval,Quebec)。人膀胱癌细胞系RT4与UMUC3购于美国典型培养物保藏中心(American Type Culture Collection)。保持细胞在含10%热灭活的胎牛血清的McCoy’s介质(Invitrogen,Burlington,ON)中并且保持在37℃下,湿润的5%CO2大气中。作为慷慨的礼物,MGHU3细胞获取自Dr.Y.Fradet(L’Hotel-DieudeQuebec,Quebec,Canada)并且保持在补充了10%胎牛血清与2mM左旋谷氨酰胺的MEM(Invitrogen)中。Dr.C.Dinney(MD Anderson Cancer Center,Houston,TX,USA)友好地提供了KU7并且保持在含5%胎牛血清的DMEM中。出于显示的目的,通过Dr.Graig Logsdon(M.D.Anderson Cancer Center,Houston,TX,USA)用含萤火虫荧光素酶基因的慢病毒感染KU7细胞,并且这些亚克隆被命名为如早先报道的KU7-荧光素酶(Hadaschik BA,Black PC,Sea JC等.BJUInt2007;100:1377-84)。
细胞以5,000个细胞/孔沉积于100μl体积的补充了10%FBS的McCoy’s介质的96孔平板中并且在添加新鲜制备的或含PTX的HPG-C8/10-MePEG和/或HPG-C8/10-MePEG-NH2(溶解于PBS中,pH7.4)溶液前允许平衡24小时。细胞暴露于药物制剂2小时以模拟目前的用于灌注治疗的临床标准并且如早先报道的(Mugabe C,Hadaschik BA,Kainthan RK等,BJU Int 2009;103:978-86),利用CellTiter96水的非放射性细胞增殖测定(MTS)(Promega,Madison,WI)在72小时后确定细胞活力。每一实验重复三次并且MTS值适合于所有细胞系的线性吸光率。
所有制剂都导致所有测试的细胞系增殖的浓度依赖性抑制。更有侵略性并且快速生长的KU7-荧光素酶细胞系对DTX制剂最敏感。发现负载DTX的HPG-C8/10-MePEG或HPG-C8/10-MePEG-NH2和商品化制剂的细胞毒性是一样的(图25)。对所有测试的细胞系而言,DTX制剂的IC50处于低的纳摩尔范围(4-12nM)。对照的HPG纳米颗粒(无药物)在测试的浓度范围内(15-1,500nM,数据未显示)没有显示出细胞毒性。HPG中DTX的负载对它的细胞毒性没有影响。
实施例21:体内研究
膀胱内DTX在膀胱癌的原位小鼠模型中的功效
在总数为42只裸鼠中完成体内研究以评估用(0.2mg/ml)及负载DTX(0.2mg/ml)的HPG-C8/10-MePEG和/或HPG-C8/10-MePEG-NH2单一膀胱内处理的功效。已报道了所用的原位小鼠模型(Hadaschik BA,Black PC,Sea JC等.BJU Int 2007;100:1377-84;Mugabe C,Hadaschik BA,Kainthan RK,等.BJU Int 2009;103:978-86;Hadaschik BA,ter Borg MG,Jackson J等.BJU Int 2008;101:1347-55;Hadaschik BA,Adomat H,Fazli L等.Clin Cancer Res 2008;14:1510-8;Hadaschik BA,Zhang K,So AI等.Cancer Res 2008;68:4506-10)。根据加拿大动物管理委员会的要求进行动物研究。用异氟烷麻醉11周大小雌性裸鼠(Harlan,Indianapolis,IN)。在润滑的24G Jelco血管探针(Medex Medical Ltd.,Lancashire,UK)穿过尿道至膀胱之前将表浅的6-0聚丙烯荷包缝合置于尿道口周围。膀胱的PBS单一冲 洗后,灌注200,0000个KU7-荧光素酶细胞作为50μl单一细胞悬浮液并且将荷包缝合结扎2.5小时。为定量体内肿瘤负载,在4,11,18以及25天腹膜内注射150mg/kg荧光素后,用IVIS200成像系统(Xenogen/Caliper Life Sciences,Hopkinton,MA)在仰卧位对动物成像15分钟。利用Living Image软件(Xenogen)获取及分析数据。在肿瘤接种后第5天,小鼠随机接受单一的50μl PBS(对照);(0.2mg/ml);负载DTX(0.2mg/ml)的HPG-C8/10-MePEG;以及负载DTX(0.2mg/ml)的HPG-C8/10-MePEG-NH2的膀胱内处理。生物荧光水平在组间是相等的,然而,因为肿瘤因个体小鼠的不同而异,为统计分析,每只小鼠中在第4天,以最初的通量做参比,将处理后的肿瘤生物荧光标准化。肿瘤接种后第25天进行尸检。移除整个膀胱,固定于10%缓冲的福尔马林并且埋入石蜡中。制备5μm切片并且利用标准的技术用H&E染色。在BLISS显微镜成像工作站(Bacus Laboratories Inc.,Lombard,IL)上评估及扫描所有的载玻片。
在KU7-荧光素酶癌细胞的膀胱内接种后,所有的小鼠发展为膀胱肿瘤。然而,负载DTX的HPG-C8/10-MePEG-NH2组中的1只小鼠在处理后第4天意外地死亡。总之,小鼠很好地耐受了作为商品化 或HPG制剂给药的膀胱内DTX并且没有观察到主要的毒性。
与对照小鼠相比,负载DTX的HPG抑制肿瘤生长。然而,负载DTX的HPG-C8/10-MePEG-NH2为最有效的制剂:抑制肿瘤在KU7-荧光素酶原位膀胱癌异种移植中生长并且与PBS对照或组相比达到统计学意义(图26,P<0.01,双因素方差分析后post-hoc Bonferoni分析)。在研究的最后,与PBS对照组相比,负载DTX的HPG-C8/10-MePEG-NH2纳米颗粒的单一膀胱内灌注抑制了88%肿瘤生长。负载DTX的HPG-C8/10-MePEG纳米颗粒在该处理组中显示了54%肿瘤抑制。功效的增加很可能源于负载DTX的HPG-C8/10-MePEG-NH2纳米颗粒处理的小鼠的膀胱及肿瘤组织中增强的药物摄取。
商品化制剂在该原位异种移植模型中不能抑制肿瘤生长。每一处理组中小鼠随时间变化的代表性生物荧光图像显示于图26中。膀胱组织的组织学检查显示KU7-荧光素酶肿瘤表现出侵略性生长 方式及频发的多灶性,但是肿瘤接种后25天,它们通常限于固有层以及与高级的T1阶段疾病关联(图27)。尽管DTX(0.2mg/ml)不引起KU7-荧光素酶异种移植中任何异常的组织学改变,与PBS处理相比,负载DTX(0.2mg/ml)的HPG-C8/10-MePEG和/或HPG-C8/10-MePEG-NH2抑制肿瘤生长。通过HPG-C8/10-MePEG-NH2中负载的DTX处理的肿瘤大小显著地减小,伴有异质细胞大小,细胞核形状以及侵润的炎性细胞。
也确定的是用于负载生物活性部分至dHPG的技术能够产生制剂,测定该制剂为递送系统中药物的靶标量的±20%,如通过HPLC所测定的(见表11)。
表11处理后通过HPLC所分析的样品中DTX制剂的浓度
在掺入紫杉醇(PTX)的制剂与掺入DTX的制剂间进行比较,如图28所示。对这两种药物而言,将它们掺入至诸如HPG-C8/10-MePEG的dHPG,导致减少的肿瘤发光,并且掺入DTX的dHPG比掺入紫杉醇的dHPG更有效。
利用两个时间点以及利用健康的动物重复相似的实验。图29中所示的这些数据显示健康小鼠中,使用HPG-C8/10-MePEG-NH2比HPG-C8/10-MePEG或TaxotereTM用于递送相同量的药物时,随着时间的变化,更多的多西紫杉醇保留在膀胱中。对HPG-MePEG-NH2而言,由于超过定量的测定最高极限,结果为半定量的。对TaxotereTM与HPG-C8/10-MePEG组而言,结果为定量的。每次给予小鼠50μL体积中的50μg药物并且使用50mg dHPG(n=3/组)。
实施例22:药物摄取研究
为评估膀胱内DTX制剂后膀胱组织与血清摄取,用(0.2mg/ml,n=3)或负载DTX(0.2mg/ml)的HPG-C8/10-MePEG(n=4)和/或HPG-C8/10-MePEG-NH2(n=4)灌注具有原位膀胱肿瘤的小鼠。灌注后两小时测量尿液、膀胱组织以及血清中DTX的量。在具有确定的KU7-荧光素酶肿瘤(肿瘤接种后33天)的15周大小雌性裸鼠中进行药物摄取研究。在膀胱内灌注后0,30以及60分钟时取尾部血液样品。在这期间仍然用异氟烷麻醉小鼠。2小时后,利用CO2窒息将所有小鼠安乐死并且通过心脏穿刺除去额外的血液。在微血细胞比容管(Fisher Scientific,Pittsburg,PA)或血清分离管(Becton Dicknson)中离心血液样品并且在液氮中快速冷冻血清。也收获每只小鼠的尿液和膀胱并且在冷冻前,将膀胱切开以暴露内腔,并且在五次连续的10ml PBS洗涤液中强力地洗涤。所有样品储存于-80℃。由集成的超高压液相色谱(Waters Acquity UPLC)分离系统组成的UPLC-MS/MS系统用于分析,结合利用Waters TQD质谱仪的质谱分析。在以下情况下操作系统:电喷射离子源阻断温度150℃,脱溶剂温度350℃,采样锥电压45kV,毛细管电压0.70kV,提取器电压3kV,RF电压0.1kV,进样锥气体流速25l/h,脱溶剂气体流速600l/h以及碰撞气体流速0.2ml/min。分子经受阳离子模式的电喷雾离子阱。在具有m/z 808.5→527.2的转变的多反应监测中定量DTX,如早先确立的(Mugabe C,Liggins RT,Guan D,et al.Int J Pharm 2011;404:238-49)。通过溶剂/溶剂提取方法从小鼠血清提取DTX。将50μl等分的小鼠血浆和标准液与150μl含0.1%甲酸的乙腈混合于96孔平板并且在室温下涡旋1分钟。在4℃下以5,500转速(AllegraTM 25R离心机,Beckman-Coulter)离心样品10分钟。然后将100μl上清液与50μl蒸馏水混合,混合并且涡旋30秒。称重膀胱组织并且在0.1%甲酸/甲醇中利用氧化锆珠(Biospec Products)及装备有微量试管架的微型珠搅拌器(Biospec Products)均质化60秒。在4℃下以14,000转速(AllegraTM 25R离心机,Beckman-Coulter)离心样品2分钟。添加150μl含0.1%三氟乙酸的甲醇至样品,混合并且在4℃下以14,000转速(AllegraTM25R centrifuge,Beckman-Coulter)涡旋15分钟。利用UPLC-MS/MS进行所有的样品分析。DTX的定量限为10ng/ml, 具有来自加入标准的对照样品97%的恢复。批内精密度(%RSD)在所有的情况下都不超过15%。
所有时间点上,灌注的小鼠在血清中都没有可检测的DTX。负载DTX的HPG-C8/10-MePEG-NH2在2h时间点上显示了最高的血清水平(150.87±34.98对23.97±16.71ng/ml,P<0.01,双因素方差分析,Bonferronipost-test)。然而,DTX的血清浓度比尿液和膀胱组织中浓度低几个数量级(表12)。与或负载DTX的HPG-C8/10-MePEG比较,负载DTX的HPG-C8/10-MePEG-NH2导致了膀胱组织积聚中明显更高的量(P<0.001,单因素方差分析,Bonferroni’s多重比较检定)。与负载DTX的HPG-C8/10-MePEG处理组在膀胱组织积聚中没有显著的差异(P>0.05,单因素方差分析)。最终的尿液浓度比最初的计量溶液低约5-7倍。这是由于膀胱内灌注2小时期间尿液的稀释。然而,不同处理组间DTX的最终尿液浓度没有显著的差异(P>0.05,单因素方差分析)。任一组中没有观察到局部或全身的毒性。
表12膀胱内DTX制剂在原位异种移植中的药物摄取
1通过HPLC所测量的膀胱内灌注2小时后小鼠尿液中DTX的最终浓度
2通过LC/MS/MS所测量的膀胱内灌注2小时后小鼠膀胱组织中DTX的浓度
3通过LC/MS/MS所测量的膀胱内灌注后在0.5、1以及2小时取的小鼠血清中DTX的浓度
BLOQ,低于定量极限(最低的定量极限为10ng/ml)
数据显示为平均值±SD
实施例23:评估肿瘤微环境与若丹明标记的HPG的摄取
评估膀胱肿瘤微环境与若丹明标记的HPG分布至肿瘤组织。
HPG的若丹明标记
如早先报道的(Savic R,Luo L,Eisenberg A,Maysinger D.Science2003;300:615-8;Mugabe C,Liggins RT,Guan D,et al.Int J Pharm 2011;404:238-49)用四甲基若丹明-5-羧基叠氮化物(TMRCA)共价地标记HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2。用异氟烷麻醉具有原位膀胱肿瘤的15周大小雌性裸鼠(肿瘤接种后33天)。将表浅的6/0聚丙烯荷包缝合置于尿道口周围并且通过人工压迫排空膀胱。将润滑的24号Jelco血管导管穿过尿道至膀胱然后灌注50μl任一PBS、游离的若丹明(TMRCA)、HPG-C8/10-MePEG-TMRCA和/或HPG-C8/10-MePEG-NH2-TMRCA,并且将荷包缝合结扎2小时,在此期间保持小鼠处于麻醉状态。2小时后移除荷包缝合,通过人工压迫排空膀胱并且用150μl PBS(pH6.0)洗涤两次。使小鼠安乐死,切离膀胱,并且将其在铝块上冷冻,然后包埋于OCT用于冰冻切割。从膀胱边缘以1、2以及3mm的距离切取10μm冰冻切片。室温下干燥切片并且利用10倍的目标(0.75μm/像素间距)以若丹明荧光对切片成像。载玻片固定于1:1的丙酮:甲醇溶液10分钟并且利用定制的毛细管作用染色器针对CD31(1:50具有抗仓鼠Alexa 647二抗的仓鼠抗-CD31)与Hoechst 33342(细胞核染料)。CD31与Hoechst 33342荧光成像后,轻轻地用苏木精复染色,亮视野中固定&成像。
图像分析:图像简化为1.5μm/像素间距以改善图像J软件中的可使用性。然后用用户提供的算法建立图像栈,比对及裁剪至肿瘤组织边界,移除处理工具;基于苏木精图像进一步地裁剪坏死。在Hoechst33342图像上沿着肿瘤组织边界人工地示踪膀胱内腔。运行用户提供的分析宏命令以产生下列的数据类型:a)阈值:人工地确定以包括阳性染色但是其并没有在坏死区域以外获得背景;宏命令确定满足或超过该阈值的正性像素的数量并且报道称之为全部肿瘤切片的平均值。b)强度:报道称之为对全部肿瘤切片染色的平均强度,或基于它们与 第二次染色(即CD31)的距离或人工示踪边界(膀胱内腔)将像素的平均强度分类。进行计算以确定平均值±标准差并且利用微软电子表格(Microsoft Excel)建立图形显示;利用用于Macs软件的棱镜v5进行方差(Kruskal-wallis检验)统计分析的非参数分析。
评估膀胱肿瘤微环境及若丹明标记的HPG至肿瘤组织的分布。膀胱肿瘤组织为高度血管吻合的,至最近的血管平均距离40-60μm(图30A)。在不同的组间没有见到显著的差异(P=0.8)。测量整个膀胱肿瘤内的荧光量。与其他的组相比(P=0.037),若丹明标记的HPG-C8/10-MePEG-NH2(HPG-C8/10-MePEG-NH2-TMRCA)显示了最高的肿瘤摄取。在游离的若丹明(TMRCA)与若丹明标记的HPG-C8/10-MePEG所灌注的膀胱的肿瘤摄取中没有显著的差异(P>0.05)(图30B)。根据膀胱内腔距离评估若丹明摄取至肿瘤组织的深度分布。HPG-C8/10-MePEG-NH2-TMRCA纳米颗粒证明了在所有内腔距离上增强的肿瘤摄取,显示了超过HPG-C8/10-MePEG-TMRCA纳米颗粒的5-6倍增加(图30C)。
实施例24:HPG-C8/10-MePEG与HPG-C8/10-MePEG-COOH的合成及特征
根据我们早先报道中所述的方案(Kainthan,R.K.;Brooks,D.E.Bioconjugate Chem.2008,19,2231–2238)进行O/DGE内核修饰的HPG的聚合作用。根据以前报道的方案(Haxton,K.J.;Burt,H.M.Dalton Trans.2008,5872–5875)进行具有羧酸基的C8/10内核-修饰的HPG的功能化。对典型的反应而言,将5.0g HPG-C8/10-OH或HPG-C8/10-MePEG6.5溶解于100ml吡啶中,并且将溶液保持在氮气下,随后添加二甲氨基吡啶与琥珀酰酐,根据HPG上羧酸基的靶标量调节。为合成HPG与最高量的COOH基,将所有可利用的游离羟基靶向用于羧化物的修饰;因此,添加过量的二甲氨基吡啶(0.075g,0.61mmol)与琥珀酰酐(4.5g,45mmol)至反应混合物。通过计算游离羟基的理论模型,确定了存在348mols游离羟基/HPG摩尔,因此,理论上,相同数量的羧基/HPG摩尔。因此,由此得到的HPG表示为HPG-C8/10-MePEG6.5-COOH348。 少量使用二甲氨基吡啶(0.015g,0.12mmol)与琥珀酰酐(0.9g,9mmol)产生了HPG,其中并不是所有的游离羟基都靶向用于修饰。通过计算添加至反应混合物的琥珀酰酐的摩尔数量确定羧酸基添加至HPG的理论数量。因此,这种含HPG的低羧化物表示为HPGC8/10-MePEG6.5-COOH113。添加二甲氨基吡啶与琥珀酰酐后,室温下使用磁性搅拌棒搅拌溶液过夜。添加去离子水(100mL)至烧瓶并且将混合物保持搅拌30分钟。通过旋转蒸发除去溶剂,伴有周期的水添加以使吡啶通过共沸蒸馏能够更好的蒸发。将最终的产物溶解于甲醇中并且以80:20甲醇/去离子水的混合物作参比,利用醋酸纤维素透析袋(MWCO 10000g/mol,Spectrum Laboratories Inc.,Rancho Domunguez,CA)透析3天,每隔8小时改变透析介质,每次用较低的甲醇浓度直至在最后的三个阶段期间,透析介质为100%水。通过冷冻干燥获得聚合物。
HPG-C8/10-MePEG-COOH的13C NMR(400MHz,甲醇-d4)δC:0(四甲基硅烷,内标准),14.73(O/DGE上的CH3,烷基),23.92-33.24(C(O)CH2CH2COOH),48.51-49.86(溶剂,甲醇-d4),59.29(CH3O-MePEG),64.19-65.36(-CH2OH,聚合物中未反应的伯醇基团),69.98-73.74(聚合物中的-CH2-O-,-CH-O),78.93-80.14(聚合物中的CH),173.84-174.16(C(O)CH2CH2COOH),175.92(C(O)CH2CH2COOH)。
在所有官能化HPG的制备中,将MePEG与COOH基团添加至反应混合物。各种官能化HPG的MePEG与COOH的靶标量总结于表13中。高的与低的羧化物官能化HPG的反应产率分别为84%与74%。通过下列的命名法描述HPG聚合物:HPG-C8/10-MePEGA-COOHB,其中HPG-C8/10烷基取代的HPG,A为与聚合物结合的MePEG的靶标含量,基于试剂的化学计量学(MePEG摩尔/TMP抑制剂摩尔)并且B为COOH的预期摩尔含量/HPG聚合物摩尔,基于来自于GPC数据所计算的聚合物的分子量。
表13一系列表面修饰的用C8/10烷基衍生的超支化聚丙三醇的特性
a命名法指定如下:HPG-C8/10-OH为“基础的聚合物”并且所有其他的都是用MePEG与COOH表面修饰,表示为反应中所添加的表面基团的理论摩尔数/HPG摩尔。b通过pH滴定所确定的COOH基团摩尔/HPG摩尔。c通过GPC确定的平均分子量与多分散性指数。通过Mw/PDI计算重均分子量数。d颗粒大小(直径),通过动态光散射技术所确定。
NMR分析
纯化后,通过NMR分析表征所有的HPG:利用400MHz Bruker Avance II+光谱仪(Bruker Corporation,Milton,ON)获得HPG聚合物的NMR光谱。将聚合物溶解于DMSO-d6或甲醇-d4(Cambridge Isotope Laboratories,Andover,MA)。获得一维的质子与碳光谱及二维的多重编辑的异核单量子相关谱(HSQC),异核多键相关(HMBC)以及HSQC-TOCSY(全相关谱)NMR实验。化学位移参考残余的溶剂峰。利用Sparky(T.D.Goddard and D.G.Kneller,Sparky 3,University of California,San Francisco)分析二维光谱。从HSQC数据估算HPG上的COOH的摩尔分数如下:对每一修饰而言,整合对应四个亚甲基质子的峰并且它的积分校正质子数。该值除以TMP甲基的积分(校正质子多样子)以产生COOH的摩尔分数。
自图31可以见到所有官能化HPG-C8/10-MePEG6.5-COOH聚合物的峰归属于HPG的结构组分并且与早先的报道一致(Kainthan,R.K.;Mugabe,C.;Burt,H.M.;Brooks,D.E.Biomacromolecules 2008,9,886–895;Kainthan,R.K.;Janzen,J.;Kizhakkedathu,J.N.;Devine,D.V.;Brooks,D.E.Biomaterials 2008,29,1693–1704;Haxton,K.J.;Burt,H.M.Dalton Trans.2008,5872–5875)。HSQC、HMBC以及HSQC-TOCSY利用整合的峰体积,用于估算HPG上取代基、C8/10烷基链、MePEG以及COOH的分数。
支化度与聚合作用的程度
利用下列的方程式(Ho¨lter,D.;Burgath,A.;Frey,H.Acta Polym.1997,48,30–35),通过支化度(DB)与聚合作用的程度(DPn)典型地表征超支化聚合物:
其中,DB为支化度,D、L13及L14分别代表树状的、直链的1-3及直链的1-4单元。缩水甘油的树状的与直链重复单元的结构总结于图32中,所述重复单元存在于超支化结构中。而且,计算这些聚合物的聚合作用的程度(DPn)如下(Sunder,A.;Hanselmann,R.;Frey,H.;Mu¨lhaupt,R.Macromolecules 1999,32,4240–4246):
其中D、L13以及L14如上文所定义的,T代表末端单元的分数,并且fc为内核分子的官能化(对TMP而言,其为3)。D由一级与二级单元的总数所给出的。Dp与Ds(见图32),以及L13、L14和T以相似的方式定义。
当2D HMBC与HSQC-TOCSY实验联合使用时,属于对应一级和二级L13、L14、T以及D单元的许多峰为未修饰的HPG聚合物(其作为参照物合成,不含C8/10烷基组分并且没有添加MePEG或羧基修饰,数据未显示),来自于多重编辑的HSQC的峰体积用于计算DB与DPn。 针对我们修饰的HPG而获得的结果为DB)0.51与DPn)14.83,并且结构单元的相对丰度,对线性单元而言为39%,对树状单元而言为20%,以及对末端单元而言为41%。这些数值与文献数值有很好地一致性(Sunder,A.;Hanselmann,R.;Frey,H.;Mu¨lhaupt,R.Macromolecules 1999,32,4240–4246;Ho¨lter,D.;Burgath,A.;Frey,H.Acta Polym.1997,48,30–35)。当比较C8/10烷基链修饰的HPG(HPG-C8/10-OH)的HSQC光谱与未修饰的HPG的HSQC光谱时,在前者的聚合物内核的光谱区域中可见两个新的峰(图33)。一个峰属于脂肪族链的R-亚甲基,而第二个峰不能明白地分配。基于化学位移,该峰可对应具有烷基链的T单元,所述烷基链与仲醇羟基结合;然而,不能证实这一推测。对HPG-MePEG6.5而言,观察到相似的情况。可分配来自MePEG的R-亚甲基的峰,但是额外的未知的峰不能明确地分配。与HPG-C8/10-OH相似,新的峰的化学位移与类L14单元相似。总之,不能从NMR数据计算DB与DPn,由于缺乏明确地信号分配。通过线性或末端单元的观察并且证实所有预期的支化模式和修饰(烷基、MePEG以及羧基)存在,NMR数据顾及到了游离羟基的直接表征。图33示例了NMR光谱中的各种分配峰,其显示了预期支化模式的存在和MePEG、烷基链以及COOH基团的存在。
COOH的摩尔分数
对所有COOH修饰的HPG聚合物而言,从HSQC NMR光谱估算COOH的摩尔分数。通过这种方法,HPG聚合物中COOH的数量不是绝对数,因为它表示为相对于TMP甲基存在于样品中。假设每个HPG分子只含有一个TMP;然而,不能独立地定量各种批次聚合物中TMP量/HPG摩尔。因此,这些数字用作有多少羟基被COOH帽化的定性指标。而且,因为从同一批次的HPGC8/10-MePEG6.5合成HPG-C8/10-MePEG6.5-COOH聚合物,所以预期TMP含量是相同的并且可以比较NMR光谱以确定COOH在这二者HPG-C8/10-MePEG6.5-COOH聚合物中的相对量。摩尔比率表明,与HPG-C8/10-MePEG6.5-COOH113相比,HPG-C8/10-MePEG6.5-COOH348中的COOH含量高2.8倍。对HPG-C8/10-COOH与高羧基密度的HPGC8/10-MePEG6.5-COOH348聚合物而言,未观察到对应于线性或末端基团的峰,这表明没有羟基存在于该聚合物中(见代表性NMR光谱的图34)。对低密度COOH聚合物,HPG-C8/10-MePEG6.5-COOH113而言,除了新的峰之外还观察到线性与末端基团的峰,这表明只有部分饱和的具有羧酸的羟基(数据未显示)。
FT-IR
利用具有通用的ATR采样配件的Perkin-Elmer FTIR光谱仪(Perkin-Elmer,Woodbridge,ON)获得HPG的FT-IR光谱。扫描范围为4000-650cm-1,具有4cm-1的分辨率。
HPG-C8/10-MePEG6.5、HPG-C8/10-MePEG6.5-COOH348以及HPG-C8/10-MePEG6.5-COOH113的FT-IR光谱显示于图35中。2800-3000cm-1处的峰与C-H振动一致并且发生在所有HPG中。1680-1780cm-1处的峰由CdO波段产生,这表明了HPG-C8/10-MePEG-COOH聚合物中COOH基团的存在。1200-1400与1000-1180cm-1处的峰分别由C-H波段与C-O振动产生,因此,可以在所有这些聚合物中发现它们。通过比较HPG-C8/10-MePEG6.5、HPG-C8/10-MePEG6.5-COOH113以及HPG-C8/10-Me-PEG6.5-COOH348的FT-IR光谱,可以见到OH峰(3300-3500cm-1)减少并且CdO峰(1680-1780cm-1)在HPG-C8/10-MePEG-COOH中出现,这表明OH基团已消耗并且转换为COOH。HPG-C8/10-MePEG6.5-COOH348的光谱显示了OH峰接近消失,这表明OH基大部分已消耗并且转换成COOH,而HPG-C8/10-MePEG6.5-COOH113的OH峰减少但是仍然明显,与NMR结果一致。而且,后者HPG也显示了较小的CdO峰,这表明与HPG-C8/10-MePEG6.5-COOH348相比COOH较低的摩尔比率。FT-IR数据显示了支持HPG的官能化改变的好证据并且也确证了通过纯化方法除去了未反应的试剂。
分子量
通过配备了DAWN-EOS多角度激光散射(MALLS)检测器与Optilab RI检测器(Wyatt Technology Inc.,Santa Barbara,CA)的凝胶渗透色谱法(GPC)(GPC-MALLS)确定HPG的重均分子量(Mw)与多分散 性。将0.1N硝酸钠水溶液用作0.8mL/min流速的流动相。早先报道中已描述了细节(Kainthan,R.K.;Brooks,D.E.Bioconjugate Chem.2008,19,2231-2238;Kumar,K.R.;Kizhakkedathu,J.N.;Brooks,D.E.Macromol.Chem.Phys.2004,205,567-573)。对各种HPG而言,确定dn/dc值在0.1N NaNO3水溶液中对HPGC8/10-MePEG6.5、HPG-C8/10-MePEG6.5-COOH348以及HPG-C8/10-MePEG6.5-COOH113而言分别为0.146、0.165以及0.138并且用于聚合物分子量的计算。利用由Wyatt Technology Corp所提供的Astra软件处理数据。通过Mw除以PDI计算聚合物的平均分子量数。
这些HPG的分子量与多分散性显示于表13中。官能化的HPG(HPG-C8/10-MePEG6.5-COOH)显示了与HPG-C8/10-MePEG6.5相比分子量增加。而且,发现在表面官能化后,聚合物的多分散性没有很大地改变,这表明了相对一致的表面修饰。分子量值与早先报道的HPG-C8/10-MePEG(Kainthan,R.K.;Mugabe,C.;Burt,H.M.;Brooks,D.E.Biomacromolecules 2008,9,886-895;Kainthan,R.K.;Janzen,J.;Kizhakkedathu,J.N.;Devine,D.V.;Brooks,D.E.Biomaterials 2008,29,1693-1704;Mugabe,C.;Hadaschik,B.A.;Kainthan,R.K.;Brooks,D.E.;So,A.I.;Gleave,M.E.;Burt,H.M.BJU Int.2009,103,978-986;Kainthan,R.K.;Brooks,D.E.Bioconjugate Chem.2008,19,2231-2238)的分子量值相似。
COOH基团的滴定
在T-50M滴定仪(Mettler Toledo,Mississauga,ON)上进行电势/pH滴定,以定量COOH表面移植的HPG的总浓度。将HPG-C8/10-MePEG6.5-COOH348与HPG-C8/10-MePEG6.5-COOH113样品溶解于0.2mg/mL10mM NaOH 10mL中。通过添加0.1M NaOH,人工升高每一溶液的pH达到大约11。然后用0.01M HCl滴定样品。注射剂设定为10-50μL的动态范围并且注射剂间30-60s的时间间隔确保平衡建立。一旦pH达到3.0就终止滴定。利用标准的外推/交叉方法确定滴定终点。报道的COOH滴定值代表三个测量的平均值。
通过电势/pH滴定(表13)测量结合HPG的COOH基团的摩尔比率 并且显示了与靶标摩尔比率及测量的分子量有很好的一致性。例如,也可通过添加具有归于COOH基团分子量的HPG-C8/10-MePEG6.5(6.3×104)的平均分子量的数量计算HPG-C8/10-MePEG6.5-COOH113的分子量,该分子量等于羧化物/HPG分子的数量(87来自于滴定数据)乘以羧化物分子量(101g/mol)。基于这种计算,HPG-C8/10-MePEG6.5-COOH113的平均分子量数为7.2×104g/mol,与测量的值有很好的一致性(7.0×104g/mol)。
溶解度
通过将聚合物已知的质量溶解于各种水缓冲液或蒸馏水评估HPG聚合物的溶解度特征。轻微地涡旋样品以加速溶解。对浑浊度的信号而言,定期地测量几天聚合物溶液在550nm处的吸光度以评估聚合物是否仍然存在溶液中。对一些羧酸衍生的HPG聚合物而言,调节溶液的pH以促进溶解。
作为潜在的药物纳米载体,这些聚合物的水介质中溶解度特征是关键的。发现HPG-C8/10-MePEG聚合物在蒸馏水、PBS缓冲液(pH7.4)以及合成的尿液中具有很好的水溶解度(大于100mg/mL)。发现HPG-C8/10-COOH在水介质或PBS(pH 7.4)中为几乎不可溶解的并且只可溶解于碱性溶液,诸如0.1M NaOH,由于在中性pH时离子化减少。HPG内核的疏水性(烷基链)组分很可能占溶解度特征的主导地位。也用MePEG基团结合的羧化物衍生的HPG显示了增加的水溶解度。因此,发现HPG-C8/10-MePEG6.5-COOH113可完全地溶解于10mM PBS,浓度为100mg/mL,没有加热,尽管溶液的pH从7.4下降至4.5。具有较高量羧化物的HPG(HPG-C8/10-MePEG6.5-COOH348)在水或PBS缓冲液中可溶解性差并且超过了缓冲作用的能力,导致了PBS缓冲液的酸化并且使pH从7.4下降至大约3.8。溶液表现出显著的浑浊度,如通过550nm处吸光度所测量的,证明了聚合物不可溶解的残余分数(数据未显示)。要求添加氢氧化钠以获得浓度100mg/mL并且澄清的溶液pH4.25。
颗粒大小与Z电势
利用Malvern NanoZS颗粒大小分析仪(Malvern Instruments Ltd., Malvern,U.K.),使用一次性定径比色皿,进行颗粒大小与Z电势分析,。在1mM NaCl pH6.0中制备浓度15mg/ml的聚合物溶液并且在测量之前用0.22μm注射器式滤器(Pall Life Sciences,Ann Arbor,MI)过滤。
羧基终止的HPG聚合物具有5-10nm范围的颗粒大小(表13)。纳米颗粒的Z电势为强烈地负性,对HPG-C8/10-MePEG6.5-COOH113与HPG-C8/10-MePEG6.5-COOH348而言,分别为-41.2±3.2与-60.3±2.1mV。Z电势中的降低是与HPG表面结合的羧基的数量的结果。
实施例25:结合HPG的顺铂
通过在0.01M NaOH中制备10mg/mL的聚合物溶液评估顺铂与羧化物修饰的HPG的结合。添加顺铂至这些溶液,从而使得药物的最终浓度范围0.5-4mg/mL。用少量的5M NaOH将每一溶液的pH调至6.0。在37℃下溶液孵育过夜,并且以50转速摇动。将溶液转移至Nanosep 3K Omega离心过滤装置(Pall Life Sciences,Ann Arbor,MI)并且以5000转速离心10分钟。用0.01M NaOH将少量过滤液(10-40μL)稀释至400μL,通过早先描述的邻苯二胺(OPDA)比色分析测定(Haxton,K.J.;Burt,H.M.Dalton Trans.2008,5872-5875)测定过滤液中未结合的顺铂的浓度。通过从添加至HPG的药物最初浓度减去过滤液中发现的未结合的顺铂的浓度确定与HPG结合的顺铂的浓度。
通过协调药物至聚合物上末端的羧化物基团获得顺铂与HPG的结合(图36)。对HPG-C8/10-MePEG6.5-COOH113而言,顺铂与聚合物的结合接近100%,效率达到最大的1mg/mL(10%w/w;图37)。在该浓度以上,在过滤液中检测到游离的药物,这表明了羧化物结合位点的饱和及介质中未结合药物的存在。在过滤液中检测到游离的药物之前,HPG-C8/10-MePEG6.5-COOH348结合达到2mg/mL,有100%效率。结合药物中的这种增加是羧化物基团数量增加的结果,因此,HPG-C8/10-MePEG6.5-COOH348上顺铂结合位点的数量与HPG-C8/10-MePEG6.5-COOH113上顺铂结合位点的数量比较。
实施例26:体外顺铂释放
顺铂与如上文所述的羧化物修饰的HPG结合,最终的聚合物浓度与顺铂浓度分别为10mg/mL与1mg/mL。添加20μL顺铂结合的聚合物溶液或1mg/mL游离的顺铂的溶液至7000MWCO Slide-A-Lyzer微型透析单元(Thermo Scientific,Rockford,IL)并且样品在37℃下透析搅拌,以4L的1mM PBS作参比,pH调至4.5、6.0以及7.4或pH7.0合成的尿液。合成的尿液(Surine)购于Dyna-Tek Industries(Lenexa,KS)。在预设的时间点上,从释放介质移除三个透析单元并且用透析单元的三次洗涤以及随后用新鲜的释放介质稀释至1mM除去全部的内容物。通过OPDA比色分析测定确定透析内容物的顺铂浓度。通过从实验开始时透析袋中最初的药物量减去残余的药物量计算释放药物的积聚百分比。数据表示为根据时间变化释放药物的积聚百分比。
游离的顺铂在PBS中的释放迅速并且在7小时内100%完全释放,这证明了膜没有在较大程度上阻碍游离药物的释放(图38)。对所有结合顺铂的HPG样品而言,发现药物以控制的方式释放,比游离药物慢相当大。PBS中,不管pH多少,与HPG-C8/10-MePEG6.5-COOH113结合的顺铂以接近相同的速率在PBS中释放,首先的2小时内,释放了大约5%,1天后释放了40%,并且7天后释放高达90%。与HPG-C8/10-MePEG6.5-COOH348结合的顺铂在pH6.0与pH7.4时以相似的速率在PBS中释放药物,接近线性方式,2小时内释放结合的顺铂大约3%,1天内释放了20%,并且7天后释放高达70%。对与HPG-C8/10-MePEG6.5-COOH348结合的顺铂而言,在pH4.5时的释放速率快于在它更高的pH时释放速率,释放属性与HPG-C8/10-MePEG6.5-COOH113的那些相似。顺铂的释放速率在尿液存在时相当地快,2小时内释放了稍微超过10%的剂量并且对HPG-C8/10-MePEG6.5-COOH113而言2天,对HPG-C8/10-MePEG6.5-COOH348而言3天,药物完全释放。与PBS中释放相似,顺铂在这两种HPG释放的差异可能是HPG-C8/10-MePEG6.5-COOH348上存在的羧化物基团数量增加的结果。因为尿液为由几种组分组成的复合混合物,不确定的是哪种化合物负责顺铂从HPG的增加式释放;然而,这种增加的释放速率可能是有利 的,提供了尿液一稀释,药物释放就增加的机制。根据顺铂从HPG的取代,可能的是尿液中含氮化合物,诸如尿素、尿酸以及肌酸酐,可与顺铂结合并且使其失活。尽管顺铂已显示了与这些化合物在一定程度上的复合,确定的是IV(静脉注射)给药后尿液中存在的大部分顺铂以最初给药的形式呈现并且高度活化的三维网状镉水解产物(Tang,X.;Hayes Ii,J.W.;Schroder,L.;Cacini,W.;Dorsey,J.;Elder,R.C.;Tepperman,K.Met.Based Drugs 1997,4,97-109)。根据这一发现,很可能的是来自尿液中HPG所释放的大部分顺铂以药理活化形式呈现。
实施例27:细胞毒性评估
利用MTS细胞增殖测定(Promega,Madison,WI)进行细胞毒性研究。该测定没有测量试剂的直接细胞溶解效应,但是测量了聚合物随长时间周期对细胞增殖的影响。在该研究中,由M.Tachibana博士(Keio University,Tokyo,Japan)友好地提供KU7-荧光素酶膀胱癌细胞。细胞以5000个细胞/孔沉积于96孔平板上180μL补充了10%胎牛血清(FBS)(Invitrogen Canada,Inc.,Burlington,ON)、1%青霉素-链霉素以及1%左旋谷氨酰胺的达尔伯克氏改良伊格尔氏培养基(DMEM)(Invitrogen Canada,Inc.,Burlington,ON)并且允许在37℃下、5%CO2中生长24小时以达到大约80%融合用于细胞毒性测定。然后将细胞与HPG单独地孵育2小时或72小时,范围为0.01-100mg/mL,或将细胞与游离的顺铂或负载顺铂的HPG单独地孵育2小时或72小时,药物浓度范围0.01-100μg/mL。处理后,用汉克斯平衡盐溶液(HBSS)洗涤细胞两次并且添加180μL新鲜的培养介质至每个孔并且允许细胞生长72小时。如早先所述的(Mugabe,C.;Hadaschik,B.A.;Kainthan,R.K.;Brooks,D.E.;So,A.I.;Gleave,M.E.;Burt,H.M.BJU Int.2009,103,978-986),利用CellTiter96水非放射性细胞增殖测定(Promega,Madison,WI)测量这些细胞的增殖。简而言之,将HBSS中180μL 10%v/v的3-(4,5-二甲基噻唑-2-基)-5-(3-羧基甲氧基酚)-2-(4-磺苯基)-2H-四唑溶液添加至每个孔并且孵育细胞2小时。利用微板阅读器,以620nm作参照,在490nm处测量吸光度。
研究孵育时间2小时与72小时无药物负载的HPG与负载顺铂的HPG对KU7-荧光素酶膀胱癌细胞的抑制作用(图39)。选择这些孵育时间考虑到模拟典型的膀胱内灌注期以及比较抗早先确定的顺铂抑制浓度。确定孵育2小时50%处的抑制浓度(IC50),对HPG-C8/10-OH、HPG-C8/10-MePEG6.5、HPGC8/10-MePEG6.5-COOH348以及HPG-C8/10-MePEG6.5-COOH113而言,分别为1.3、45.7、47.0以及63.0mg/mL。当使用72小时孵育时,聚合物抑制细胞增殖的程度大于2小时孵育时所发现的聚合物抑制细胞增殖的程度。HPG-C8/10-OH与HPG-C8/10-MePEG6.5分别具有0.1与0.2mg/mL IC50。对羧化物修饰的HPG而言,IC50减少了大约10倍,然而,这些聚合物仍然显示了高度的细胞相容性,IC50值大约5mg/mL。HPG-C8/10-MePEG与HPG-C8/10-MePEG-COOH的总体极好的生物相容性很可能起因于MePEG表面已知的细胞相容性,确保与细胞的浆膜很少的相互作用。羧酸化作用增加的益处起因于这一部分在pH7.4的网状负电荷,建立了与带负电荷的细胞表面轻微的排斥力。72小时孵育后,游离的顺铂抑制KU7-荧光素酶细胞增殖,IC50为1μg/mL(图40A),与早先对这一药物和细胞组合的报道一致(Hadaschik,B.A.;ter Borg,M.G.;Jackson,J.;Sowery,R.D.;So,A.I.;Burt,H.M.;Gleave,M.E.BJU Int.2008,101,1347-1355)。伴随2小时孵育,该IC50值增加至大约10μg/mL(图40B)。当与HPG-C8/10-MePEG6.5-COOH聚合物结合时,顺铂的复合形式也抑制了KU7-荧光素酶增殖,2小时孵育时观察到更高的IC50(大约50μg/mL),与72小时孵育值相比(大约5μg/mL)。无疑地,对2小时孵育与72小时孵育而言,顺铂的复合形式抑制细胞增殖少于游离的药物几乎5倍。与HPG复合的药物的IC50的这一增加很可能由于药物从聚合物的缓慢释放速率。
实施例28:预处理或没有预处理时,来自不同制剂的DTX与丝裂霉素F渗透至猪膀胱组织
评估来自DTX制剂的DTX与来自丝裂霉素F制剂的丝裂霉素F渗透至猪膀胱组织。将新鲜切离的猪膀胱组织切片置于Franz扩散细 胞并且用Tween 80、HPG-C8/10-MePEG或HPG-C8/10-MePEG-NH2中制备的抗癌药物DTX处理2小时。在一些实验中,用壳聚糖溶液(没有药物)或HPG-C8/10-MePEG-NH2溶液(没有药物)预处理猪膀胱组织1小时,在用Tween 80、HPG-C8/10-MePEG或HPG-C8/10-MePEG-NH2中制备的DTX处理前。对丝裂霉素F渗透研究而言,将新鲜切离的猪膀胱组织切片置于Franz扩散细胞并且用抗癌药物丝裂霉素F制剂处理2小时。在用丝裂霉素F制剂处理前,用HPG-C8/10-MePEG-NH2溶液(没有药物)预处理猪膀胱组织1小时。获得组织浓度与组织深度剖面图并且从曲线下面积(AUC)计算获得药物暴露量。
HPLC等级的乙腈与二氯甲烷获取自Fisher Scientific(Fairlawn,NJ)。液体闪烁流体CytoScintTMES,购于MP Biomedicals(Irvine,CA)。台氏(Tyrode)盐购于Sigma-Aldrich(St.Louis,MO)(台氏液包含下列以g/L为单位的成分:NaCl8.0,KCl0.3,NaH2PO4.5,H2O0.093,KH2PO40.025,NaHCO31.0,葡萄糖2.0)。多西紫杉醇获取自Natural Pharma(Langley BC.Canada)。商品化20mg/0.5mL(Sanofi Aventis,Laval,QC)购于温哥华市公立医院的BC癌症代理处。乙醇中氚标记的DTX购于Moravek Biochemicals(Brea,CA),具有23.2Ci/mmol的特异活性。通过修改实施例10中所述的方案制备HPG-C8/10-MePEG并且通过修改实施例18中所述的方案制备HPG-C8/10-MePEG-NH2。壳聚糖由Novamatrix FMC供应。猪膀胱购于Britco Inc.(Langley,BC)。就地从6-10月龄90-113kg重量的公猪上移除新鲜切离的含尿膀胱。
利用不同的方法,包括正向滴定方法、反向滴定方法以及荧光胺测定(表14),测量HPG-MePEG-NH2聚合物中每摩尔HPG-MePEG-NH2中胺的摩尔。
表14胺的摩尔/摩尔HPG-MePEG-NH2测量
a正向滴定方法-HCl作参比滴定
b反向滴定方法-添加已知量的HCl后NaOH作参比滴定
c用荧光胺衍生作用后荧光定量(实施例15中所述的荧光胺测定)
负载DTX的HPG-C8/10-MePEG、HPG-C8/10-MePEG-NH2以及Tween 80制剂的制备
利用溶剂蒸发技术,制备负载DTX的HPG-C8/10-MePEG与HPG-C8/10-MePEG-NH2。将DTX与HPG-C8/10-MePEG或HPG-C8/10-MePEG-NH2溶解于乙腈并且在60℃下烘箱中干燥1小时,用氮气闪耀以除去有机溶剂的痕迹。干燥前,将聚合物/药物溶液与小等份的3H DTX形成峰值。由此得到的聚合物/药物基质与60℃台氏缓冲液(pH7.4)重组并且涡旋2分钟。药物的最终浓度为0.5mg/mL并且在37℃下使用。通过用台氏缓冲液稀释浓缩溶液(含40mgDTX与1040mgTween 80/mL)以产生最终浓度为0.5mg/mL DTX,在Tween 80中制备DTX。稀释前将少量的3H DTX掺入溶液。
丝裂霉素F制剂的制备
在台氏缓冲液中制备丝裂霉素F(MW=363.4)。它来自于American Radiolabeled Chemicals Inc(St Louis,MO)Cat#ART-1689。活性为1-10Ci/mmol,乙醇中1mCi/mL。通过将50μL乙醇储备液溶解于3mL缓冲液,300x稀释液。
用于用作预处理的壳聚糖溶液与HPG-C8/10-MePEG-NH2溶液的制备
用于制备壳聚糖溶液的壳聚糖为PROTASANTM UP CL 213(Product#:4210106)并且基于其中75-90%的鲸蜡基移除的壳聚糖。阳离子的聚合物为高度纯化的并且以可溶于水的氯化盐为良好特征。通常,PROTASANTM UP CL 213的分子量在150000-400000g/mol范围(作为壳聚糖醋酸盐所测量的)。通过将它溶解于水中以使溶液浓度为0.5%w/v制备壳聚糖溶液。
通过将HPG-C8/10-MePEG-NH2溶解于乙腈制备用于用作预处理的HPG-C8/10-MePEG-NH2溶液。60℃下将由此得到的溶液在烘箱中干燥1小时并且用氮气闪耀以清除有机溶剂的痕迹。由此得到的聚合物与60℃台氏缓冲液(pH 7.4)重组并且涡旋2分钟。
组织制备
除去新鲜切离的猪膀胱外壁上过量的脂肪组织并且纵向地切开左外侧与右外侧,在37℃下充溢着氧和5%二氧化碳的混合气(95%O2/5%CO2)的台氏缓冲液浅浴中将其切成大约2cm x 2cm的碎片。在处死后5小时内进行所有研究。将膀胱碎片置于Franz扩散细胞装置上,从而使得膀胱壁的腔侧面暴露于药物溶液。这些组织切片没有拉伸并且测量大约2-3mm厚。受体腔内充满了10mL 37℃的台氏缓冲液(pH7.4)。修剪扩散细胞的视野计周围过量的组织。扩散细胞的供体腔充满了1mL0.5mg/ml的药物溶液并且组织暴露面积为0.64cm2。将每一扩散细胞设置在浅水浴中并且在37℃下孵育2小时。对一些实验而言,在用负载DTX或丝裂霉素F的制剂处理前,用壳聚糖溶液(没有药物)或HPG-C8/10-MePEG-NH2溶液(没有药物)对组织样品预处理1小时。用台氏缓冲液洗涤组织样品三次以除去所有未结合的药物。修剪组织样品并且用干冰床上的液态氮在金属板上快速冰冻。
组织的低温切片机切片
用Shandon CryomatrixTM(Themo Scientific,Pittsburgh,PA)将冰冻的膀胱组织置于低温切片机的载物架上。在-20℃下,在具有R404A冷冻系统的Shandon电子的低温切片机(Cryotome Electronic)(Thermo Electron Corporation,Cheshire,England)上,用Shandon MB35Premier Low Grade Microtome Blades(Themo Scientific,Pittsburgh,PA)将膀胱组织切片。将组织切片。将组织切片置于预先称重的1.5mL微量离心管中并且冰冻储存在-20℃。
组织中药物的定量
将200μL乙腈添加至称重的组织薄片用于药物提取。涡旋样品直至所有的组织薄片浸入乙腈中并且在室温下保持24小时以确保药物 的完全提取。将包括所有组织薄片的提取的样品转移至闪烁小瓶并且添加5mL闪烁液。通过液体闪烁计数测量3H DTX或3H丝裂霉素F的计数并且利用来自于最初的储备液的校准图定量。
组织的水平-深度剖面图的分析
分析组织的水平-深度剖面图用于平均的DTX或丝裂霉素F浓度,所述浓度为膀胱壁向下至肌肉的所有层,例如尿路上皮、固有层以及肌层。组织层中发现的药物的总量除以那一层的总组织重量确定平均组织水平。利用线性梯形规则计算组织-水平深度剖面图下的面积(AUC),如下:
其中,t为组织深度以μm为单位,并且C为浓度以μg/g为单位。
通过在0μm处药物浓度(μg/g)的外推获得估算以便计算AUC。
没有预处理时,DTX从DTX制剂渗透至猪膀胱组织的数据显示于图41中。dHPG评估含胺导致至约1500μm的深度更高的药物浓度,与没有胺的制剂相比,包括Tween 80制剂和不包含官能性胺的dHPG。观察到胺添加的效应为浓度依赖性的。图41显示,利用含最高的胺含量的dHPG制剂(37mol/mol),尤其是在约1500μm的深度获得组织中最高的药物浓度。已计算了在标明的组织深度范围处AUCs(表15a与15b),显示dHPG制剂在Taxotere处改善范围1.3-2.4倍。已计算了在标明的组织深度范围处Cavg与Cmax值(表15c)。
表15a没有预处理各种媒介中递送多西紫杉烷的渗透所计算的AUC(180-2640μm)
表15b没有预处理各种媒介中递送多西紫杉烷的渗透所计算的AUC(180-1560μm)
表15c对没有预处理各种媒介中递送多西紫杉烷的渗透而言,180-2640与180-1560组织深度(μm)的范围所计算的Cavg与Cmax值
来自用壳聚糖或HPG-C8/10-MePEG-NH2预处理的DTX制剂的DTX渗透至猪膀胱组织的数据显示于图42与43。实验的目的是从壳聚糖辨别dHPG聚合物的功能,所述壳聚糖也是包含胺功能的聚合物。已关注壳聚糖作为聚合物用于膀胱内递送;然而,在预处理以促进药物摄取至组织的上下文中,证明了某些组合物的dHPG为优越的。数据显示,用壳聚糖预处理,Tween 80制剂提供了相对少的组织渗透,提供多西紫杉醇在膀胱中所有组织深度最低的组织浓度。相似地,壳聚糖预处理导致了多西紫杉醇在组织渗透中适度的改善,当药物以HPG-C8/10-MePEG(没有胺)媒介给药时,然而这只是优于Taxotere处理的(壳聚糖预处理)组。当dHPG包含胺(37mol胺/聚合物mol)(图42与43中称为HPG-C8/10-MePEG-NH2)时,观察到多西紫杉醇组织渗透中的优越效应。同样地,壳聚糖对递送没有提供益处,当它用作负载多西紫杉醇的HPG-C8/10-MePEG-NH2制剂的预处理时。通过测量随组织深度变化的(图42)组织浓度的曲线下面积(AUC,图43)比较每个的性能,其为总药物暴露量的测量。结果显示当预处理或处理利用含胺的dHPG时,获得最大的暴露量。针对180-3360组织深度(μm)范围所计算的AUC值,针对180-1560与180-3360组织深度(μm)范围所计算的Cavg与Cmax值,以及各种预处理方案后来自于各种制剂的多西紫杉醇的渗透显示于表15d和15e中。
表15d各种预处理方案后用各种媒介递送多西紫杉醇的渗透所计算的AUC(180-3360μm)
表15e各种预处理方案后用各种媒介递送多西紫杉醇的渗透180-1560与180-3360组织深度(μm)范围所计算的Cavg与Cmax(180-3360μm)
用HPG-C8/10-MePEG-NH2预处理,丝裂霉素F渗透至猪膀胱组织的数据显示于图44中。
猪膀胱离体渗透研究的扫描式电子显微镜(SEM)图像
上文实施例28中所观察的药物渗透效应与暴露于各种治疗后膀胱组织的出现有关。对这些实验而言,没有使用药物并且使用单一的暴露于单一的媒介/膀胱。2小时暴露时间后,收获膀胱组织,用缓冲液漂洗并且用4%多聚甲醛及2%戊二醛固定过夜,用1%OsO4后固定,乙醇中脱水,临界点干燥。将整个膀胱分成两个并且用金喷射涂。通过SEM(Hitachi S4700,3-5kV)检查全部的表面并且记录代表性图像(最小值3个视野/样品)。显示代表性图像(大约130μm宽x100μm高)。
SEM图像展现只用缓冲液处理的膀胱的尿路上皮以及用HPG-C8/10-MePEG(没有胺)处理的膀胱显示了完整的或大部分完整的尿路上皮,例如没有雨伞细胞的缺失。相比之下,暴露于壳聚糖预处理媒介后,膀胱的表面在外观上非常不同,尿路上皮缺失了雨伞细胞的顶层(图45)。暴露于具有10mol胺/HPG mol的溶液浓度1与10%w/v的HPG-C8/10-MePEG-NH2后,观察到雨伞细胞从尿路上皮部分的缺失(图46)。相比之下,当使用具有较高胺含量的HPG-C8/10-MePEG-NH2(37mol/mol)时,观察到效应为浓度依赖性的。0.1%w/v的溶液浓度导致了膀胱表面外观很少或没有改变,而在用具有浓度分别为1%w/v与10%w/v的溶液处理后观察到雨伞细胞部分及完全的缺失(图46)。不受理论约束,据信胺效应可以改变膀胱的表面,影响其至药物渗透性的改变。效应显示为胺含量与聚合物浓度依赖性的。
实施例29:小鼠膀胱的体内渗透研究
评估不同的制剂(没有药物)暴露对小鼠膀胱的影响。利用4%异氟烷与2L/min O2麻醉8-11周大小雌性无胸腺裸鼠(Harlan,Indianapolis,IN)至深层面。膀胱完全地表达并且经由外科植入的导管灌注制剂。在将润滑的24号Jelco血管导管(BDickenson)穿过尿道至膀胱之前将聚丙烯荷包缝合置于尿道口周围。注射50μL体积,将动物轻微地反转(颅侧地)并且当迅速地移除导管时结扎荷包缝合。将小鼠保持麻醉状态(1.5-2%异氟烷2L/min O2)直至完成2小时灌注。去除荷包缝合并且允许动物恢复过来。
制备的计量溶液为无色澄清的至轻微琥珀色的溶液。聚合物溶液浓度为1%w/v或10%w/v。使用没有胺内容物的HPG-MePEG聚合物以及两种具有8-10mol(低的)与37mol(高的)胺/HPG摩尔(基于所谓的HPG分子量65kg/mol)的HPG-MePEG-NH2聚合物。计量浓度结果总结于表16中。
表16制剂浓度总结
意指没有检测
从每只小鼠切除膀胱用于SEM与组织学研究。给药后观察所有的 动物2小时并且在之前采集每一组织用于死亡率和发病率研究。没有记录到死亡率或发病率的迹象,除了在灌注部位一些少量的血液之外。
SEM分析
用PBS洗涤组织3次,用2%多聚甲醛固定过夜然后转移至0.1M卡可酸盐缓冲液。室温下用1%四氧化锇后固定组织1小时,然后在乙醇中脱水,与水混合,增加乙醇在混合物中的百分比,开始为30,增加至100%。通过临界点脱水干燥样品,然后用钯金喷涂两次(一次在90°,第二次时间在45°)。用Bioimaging Facility Hitachi S4700扫描式电子显微镜检测样品。将每一膀胱显示于低放大率然后在高放大率下再次检测全部的表面。在各种放大率下进行多次检测(9-10图像)。
表17制剂浓度总结
意指不可应用的。意指没有检测。
完整的:没有雨伞细胞缺失的迹象
+:非常少的或部分的雨伞细胞的缺失
++:实质的或完全的雨伞细胞的缺失
用PBS 2小时灌注处理的小鼠膀胱表面具有完整的雨伞细胞层并且具有如图47中小鼠膀胱表面的SEM图像所见的折叠的外观。用10%w/v HPG-MePEG溶液2小时灌注处理的小鼠膀胱表面在2小时灌注期后就具有完整的雨伞细胞层。在HPG-MePEG溶液灌注后6小时与24小时也观察到完整的雨伞细胞层(图48)。如图48所示,表面的细胞具有扁平的外观。用10%w/v HPG-MePEG-NH2(10mol/mol)溶液2小时灌注处理的小鼠膀胱表面在2小时灌注期后就具有完整的雨伞细 胞层(图49)。10%w/v HPG-MePEG-NH2(10mol/mol)溶液灌注后6小时时,小鼠膀胱表面表现出单一的雨伞细胞的缺失,暴露了上皮的底层。相同的灌注后24小时时,小鼠膀胱表面具有完整的雨伞细胞表面层(图49)。用1%w/v HPG-MePEG-NH2(37mol/mol)溶液2小时灌注处理的小鼠膀胱表面表现出雨伞细胞的完全缺失,如图50所示2小时灌注期后就暴露了上皮的底层。1%w/v HPG-MePEG-NH2(37mol/mol)溶液灌注后6小时时,小鼠膀胱表面仍然表现出雨伞细胞实质的缺失。1%w/v HPG-MePEG-NH2(37mol/mol)溶液灌注后24小时时,小鼠膀胱表面具有部分地完整的表面(图50C的上部分),伴有雨伞细胞显著的缺失(图50C的左下角)。10%HPG-MePEG-NH2(37mol/mol)溶液2小时灌注处理的小鼠膀胱表面表现出雨伞细胞的完全缺失,如图51所示2小时灌注期后就暴露了上皮的底层。10%HPG-MePEG-NH2(37mol/mol)溶液灌注后6小时时,小鼠膀胱表面也表现出雨伞细胞的完全缺失。灌注后24小时时,小鼠膀胱表面具有完整的雨伞细胞层,然而,表面的细胞显得比其他观察到的完整层更小并且在外观上更扁平(图51)。观察到制剂对雨伞细胞缺失的影响取决于dHPG的胺含量与dHPG浓度。
组织学
在紧密连接的改变、细胞的剥落以及炎性细胞的侵润方面评估组织。组织学分析结果总结于表18中。如自组织学结果中所可以见到的,在暴露于dHPG制剂后,没有在小鼠膀胱表面观察到炎症与坏死的迹象。
表18小鼠膀胱表面的组织学
尿液分析
在处死时收集尿液。在将动物安乐死后,暴露膀胱并且通过膀胱穿刺与穿过25G针的撤回除去它的内容物。尿液储存于冰上(但是没有冰冻)并且运输用于评估。用细胞的存在分析尿液。将几滴尿液置于显微镜载玻片上并且通过显微镜观察细胞的存在。用血球计载玻片计数任何的细胞存在。在2小时灌注后以及在膀胱收获的时候(2,6,24小时)就自小鼠收获的尿液中细胞计数显示于图52中。
血液分析
通过CO2吸入终止,通过心脏穿刺在最后一次呼吸后就收集血液,将大约500μL-700μL置于乙二胺四乙酸(EDTA)采血管中。将每只管反转几次以确保血液与EDTA均匀地混合以防止凝固。将血液样品储存在冰上直至对特定的时间点而言收集全部的样品然后处理以产生血浆。通过在4℃下以2500转速将样品离心15分钟(转速基于BeckmanGH 3.8A转子,RCFavg 200xg)。用移液器移取血浆上清液,将其置于标记小瓶中并且储存在-80℃。利用MesoScale平台与标准的检测试剂盒,用TNFα水平分析血液。在用各种制剂灌注后2、6以及24小时时小鼠血液中循环的TNFα水平显示于图53中。在任何样品中都没有检测到TNFα。
尽管本文公开了发明的各种实施方案,可根据本领域技术人员的 一般常识在本发明的范围内做出许多改变与修饰。此类修饰包括对发明的任何方面而言,已知的等同物的取代,为获得相同的结果以基本上相同的方式。数值范围包括界定该范围的数字。本文所用的词语“包括(comprising)”如任一开放式的术语,基本上等同于短语“包括,但不限于”,并且词语“包括(comprises)”具有相应的意义。如本文所用的,单数形式“a”,“an”以及“the”包括复数指示物,除非上下文明确另有规定。因此,例如提及“a thing”包括不止一个此类东西。
本文参考文献的引用并不承认此类参考文献为优先于本发明的技术或它构成任何承认这些文件的内容或日期。任何优先权文件以及所有的出版物,包括但不限于专利与专利申请,通过参考将本说明书中所引用的并入本文,如同每篇出版物特别地并且单独地指出通过引用并入一样如同在本文完全地列出。本发明包括基本上如上文中所述的以及关于实施例和附图的所有实施方案以及变体。
- 还没有人留言评论。精彩留言会获得点赞!