一种抗肿瘤药物的晶型化合物及其制备方法与流程
本发明属于医药
技术领域:
,涉及一种抗肿瘤药物的晶型化合物及其制备方法,具体涉及一种奈拉滨晶型化合物及其制备方法。
背景技术:
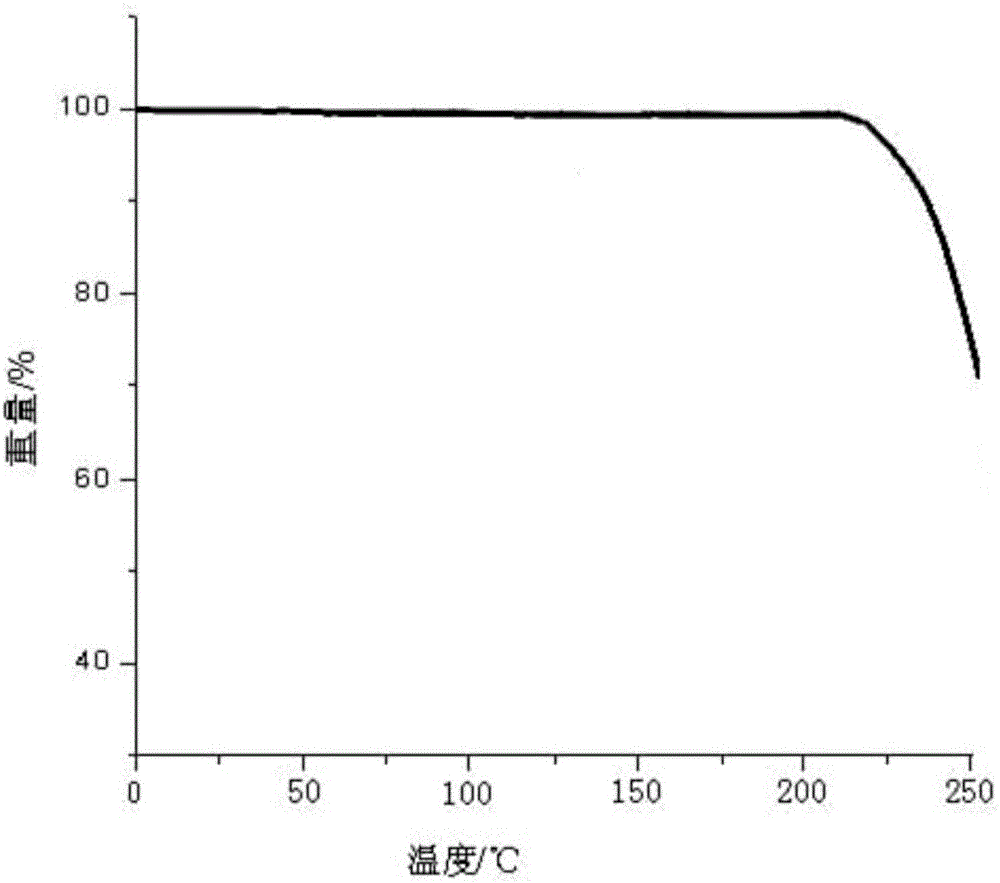
:奈拉滨化学名为2-氨基-9-β-D-阿拉伯呋喃糖基-6-甲氧基-9H-嘌呤,是脱氧鸟苷类似物9-β-D-阿糖呋喃糖鸟嘌呤(ara-G)的前体药,2005年已被批准上市,可用于治疗T细胞急性淋巴细胞性白血病(T-ALL)和T细胞淋巴母细胞性淋巴瘤(T-LBL)。结构式如下:奈拉滨为白色或类白色结晶性粉末,微溶于水,在水中溶解度约为8-9mg/ml(25℃,pH=4~10)。由于奈拉滨在水中的溶解性较差,这在一定程度上限制了其在医药上的应用。“奈拉滨的合成研究进展”【梁平,尹先清,李卫佳.精细化工中间体,2008,38(5):8-10】、CN101348511A、CN101092441A均公开了一种将奈拉滨加入甲醇混合加热至回流,搅拌使物料溶解,趁热过滤,滤液浓缩,搅拌下加入无水乙醇、乙醚,冷却放置析晶的方法。专利CN101348511A公开了奈拉滨的合成及精制,其中奈拉滨结晶的制备方法包括:奈拉滨加入甲醇混合加热至回流,溶解后趁热过滤,滤液浓缩,搅拌下加入乙醇或乙醚,放置冷却析晶,过滤,用无水乙醇洗涤,干燥,得到奈拉滨结晶,该结晶具有图3所示的X-射线粉末衍射图谱。专利CN103172687A公开了一种奈拉滨结晶化合物及其制备方法,该结晶化合物采用X-射线衍射测定法测定,得到的X射线粉末衍射图谱如图4所示,其制备方法为:取奈拉滨粗品,投入无水甲醇中,加热至回流;奈拉滨粗品溶解后加入活性炭回流,趁热过滤,降至室温,冰盐浴降温,析晶,抽滤,冰甲醇洗涤滤饼得到奈拉滨结晶化合物。专利CN103191051A公开了一种奈拉滨注射液组合物及其制备方法,其药物活性成分为奈拉滨和氯化钠,其中,所述奈拉滨为一种奈拉滨结晶化合物,该结晶化合物采用粉末X-射线衍射测定法测定,得到的X射线粉末衍射图谱如图5所示。本领域技术人员都知道,药物的多晶形已经成为药物研究过程和药品生产质量控制及检测过程中必不可少的重要组成部分。对药物多晶形的研究有助于新药化合物生物活性的选择,有助于提高生物利用度,增进临床疗效,有助于药物给药途径的选择与设计,以及药物制剂工艺参数的确定,从而提高药品生产质量。同一药物晶形不同,其生物利用度可能差异显著。同一种药物,某些晶形可能比其他晶形具备更高的生物活性。我们经过不断的研究改进,在进行了大量的试验后,通过改变结晶条件,制得了一种奈拉滨晶型化合物,并通过X-射线粉末衍射分析,证实其是一种不同于现有技术的奈拉滨的晶型化合物。技术实现要素:本发明的目的在于提供一种不同于现有技术的奈拉滨晶型化合物,该奈拉滨晶型化合物生物利用度高、稳定性好、收率高、纯度高。为实现本发明的目的,本发明采用如下技术方案:本发明所述的抗肿瘤药物的晶型化合物为奈拉滨,其以2θ±0.2°衍射角表示的X-射线粉末衍射图谱在1.769°、2.997°、4.245°、5.341°、7.121°、7.854°、12.057°、12.937°、15.958°、21.989°、35.161°、36.556°处显示有特征衍射峰。本发明所述的晶型化合物使用Cu-Kα射线测量得到的X-射线粉末衍射谱图如图1所示。该化合物的纯度达到99.5%以上,最大单杂小于0.05%。奈拉滨是一种白色或类白色结晶性粉末,CN101348511A公开了一种图3所示的X-射线粉末衍射图谱。CN103172687A公开了一种图4所示的X-射线粉末衍射图谱。专利CN103191051A公开了一种图5所示的X-射线粉末衍射图谱。研究表明,在X-射线粉末衍射图谱中,由新晶型得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。经比较发现,本发明所提供的奈拉滨晶型化合物的粉末X-射线衍射图谱(图1)与现有技术(图3、图4、图5)具有明显不同的峰的相对位置,可见其是一种与现有技术不同的晶型化合物。本发明的另一目的在于提供一种简单温和易操作,适合工业化大生产的奈拉滨晶型化合物的制备方法。本发明所述奈拉滨晶型化合物的制备方法,具体步骤如下:(1)将奈拉滨粗品加入去离子水中,加热回流,形成60~80℃下的饱和溶液;(2)加活性炭,过滤,滤液升温至90℃;(3)搅拌的条件下向滤液匀速滴加丙酮乙醚混合溶液;(4)滴加完毕后,搅拌降温,静置析晶,过滤,得奈拉滨晶型化合物湿品;(5)将步骤(4)中得到的奈拉滨晶型化合物湿品真空烘干,烘干后在惰性气体保护下破除真空冷却至室温,得到高纯度奈拉滨晶型化合物成品。优选地,所述步骤(3)中在搅拌的条件下向滤液匀速滴加的丙酮乙醚混合溶液体积为奈拉滨粗品重量的2~4倍,丙酮与乙醚的体积比为1:2~5;滴加时间控制为20~30min;滴加时的搅拌速率为15~20rmp。进一步优选地,所述步骤(3)中在搅拌的条件下向滤液匀速滴加的丙酮乙醚混合溶液体积为奈拉滨粗品重量的3倍,丙酮与乙醚的体积比为1:3.5;滴加时间控制为25min;滴加时的搅拌速率为18rmp。优选地,所述步骤(4)中的搅拌降温为梯度降温,即在转速10~20rmp下搅拌15min降温至20~30℃,再在转速10~20rmp下搅拌15min降温至-10~0℃;析晶时间为15~20小时。进一步优选地,所述步骤(4)中的搅拌降温为梯度降温,即在转速20rmp下搅拌15min降温至25℃,再在转速10rmp下搅拌15min降温至-5℃;析晶时间为18小时。优选地,所述步骤(5)中的真空烘干的温度为30~110℃。进一步优选地,所述步骤(5)中的真空烘干的温度为60~80℃。本发明进一步通过实施例和实验例,惊喜地发现本发明的奈拉滨晶型化合物较现有技术相比提高了水溶性,且生物利用度高、稳定性好、收率高、纯度高,在一定程度上拓宽了其在医药上的应用。与现有技术相比,本发明具有如下优点:(1)本发明所提供的奈拉滨晶型化合物是一种不同于现有技术的奈拉滨晶型化合物,该晶型能显著提高奈拉滨在水中的溶解度;(2)本发明所提供的奈拉滨晶型化合物具有生物利用度高、药效显著、稳定性好、收率高,纯度高等优点,有助于药物给药途径的选择设计以及药物制剂工艺参数的确定,从而提高药品生产质量;(3)本发明所提供的奈拉滨晶型化合物的制备方法操作简便,是一种经济可行、适合工业化大生产的方法。附图说明图1为本发明实施例1制备的奈拉滨晶型化合物的X-射线粉末衍射图谱;图2为本发明实施例1制备的奈拉滨晶型化合物的热分析图谱;图3为专利CN101348511A实施例19制备的奈拉滨结晶的X-射线粉末衍射图谱;图4为专利CN103172687A实施例制备的奈拉滨结晶化合物的X-射线粉末衍射图谱;图5为专利CN103191051A实施例1制备的奈拉滨结晶化合物的X-射线粉末衍射图谱。具体实施例以下用实施例对本发明的技术方案进行了详细说明,将有助于对本发明的技术方案的优点、效果有更进一步的了解,实施例不限定本发明的保护范围,本发明的保护范围由权利要求来决定。实施例中所述的奈拉滨粗品为市售的奈拉滨原料药。实施例1(1)将奈拉滨粗品100g加入去离子水中,加热回流,形成80℃下的饱和溶液;(2)加活性炭,过滤,滤液升温至90℃;(3)搅拌速率18rmp下于25min向滤液匀速滴加奈拉滨粗品重量的3倍体积的丙酮乙醚(丙酮:乙醚=1:3.5)混合溶液;(4)滴加完毕后,在转速20rmp下搅拌15min降温至25℃,再在转速10rmp下搅拌15min降温至-5℃,静置析晶18小时,过滤,得奈拉滨晶型化合物湿品;(5)将步骤(4)中得到的奈拉滨晶型化合物湿品在65℃真空烘干,烘干后在惰性气体保护下破除真空冷却至室温,得到高纯度奈拉滨晶型化合物成品95.02g,收率95.02%,光学纯度99.87%。将所制得的奈拉滨晶型化合物用美国Perkin-Elmer公司PE2400Ⅱ元素分析仪进行分析测定:元素分析(%)计算值:H5.0860%、C44.4440%、N23.5592%、O26.9108%。元素分析(%)测定值:H5.0769%、C44.4493%、N23.5586%、O26.9152%。元素分析结果显示:测定值与计算值基本一致。采用卡尔·费休水分测定法测定KF为0.39%,所以本发明奈拉滨晶型化合物不含结晶水。所制得的奈拉滨晶型化合物以2θ±0.2°衍射角表示的X‐射线粉末衍射图谱在1.769°、2.997°、4.245°、5.341°、7.121°、7.854°、12.057°、12.937°、15.958°、21.989°、35.161°、36.556°处显示有特征衍射峰,使用Cu‐Kα射线测量得到如图1所示的X射线粉末衍射图谱。与现有技术各晶型的X‐射线粉末衍射图谱进行仔细对比,明显发现本发明奈拉滨晶型化合物不同于现有技术。将所制得的奈拉滨晶型化合物采用美国Perkin‐Elmer公司PEPyrisDiamondTG热分析仪得到的热重分析图如图2所示。实施例2(1)将奈拉滨粗品100g加入去离子水中,加热回流,形成70℃下的饱和溶液;(2)加活性炭,过滤,滤液升温至90℃;(3)搅拌速率20rmp下于20min向滤液匀速滴加奈拉滨粗品重量的2倍体积的丙酮乙醚(丙酮:乙醚=1:2)混合溶液;(4)滴加完毕后,在转速10rmp下搅拌15min降温至30℃,再在转速20rmp下搅拌15min降温至-10℃,静置析晶15小时,过滤,得奈拉滨晶型化合物湿品;(5)将步骤(4)中得到的奈拉滨晶型化合物湿品在60℃真空烘干,烘干后在惰性气体保护下破除真空冷却至室温,得到高纯度奈拉滨晶型化合物成品94.49g,收率94.49%,光学纯度99.88%。将所制得的奈拉滨晶型化合物用美国Perkin-Elmer公司PE2400Ⅱ元素分析仪进行分析测定:元素分析:元素分析(%)计算值:H5.0860%、C44.4440%、N23.5592%、O26.9108%。元素分析(%)测定值:H5.0871%、C44.4461%、N23.5586%、O26.9082%。元素分析结果显示:测定值与计算值基本一致。采用卡尔·费休水分测定法测定KF为0.41%,所以本发明奈拉滨晶型化合物不含结晶水。所制得的奈拉滨晶型化合物使用Cu‐Kα射线测量得到的X‐射线粉末衍射谱图与实施例1相似;采用美国Perkin‐Elmer公司PEPyrisDiamondTG热分析仪得到的热重分析图与实施例1相似。实施例3(1)将奈拉滨粗品100g加入去离子水中,加热回流,形成60℃下的饱和溶液;(2)加活性炭,过滤,滤液升温至90℃;(3)搅拌速率15rmp下于30min向滤液匀速滴加奈拉滨粗品重量的4倍体积的丙酮乙醚(丙酮:乙醚=1:5)混合溶液;(4)滴加完毕后,在转速15rmp下搅拌15min降温至20℃,再在转速15rmp下搅拌15min降温至0℃,静置析晶20小时,过滤,得奈拉滨晶型化合物湿品;(5)将步骤(4)中得到的奈拉滨晶型化合物湿品在80℃真空烘干,烘干后在惰性气体保护下破除真空冷却至室温,得到高纯度奈拉滨晶型化合物成品94.21g,收率94.21%,光学纯度99.86%。将所制得的奈拉滨晶型化合物用美国Perkin-Elmer公司PE2400Ⅱ元素分析仪进行分析测定:元素分析:元素分析(%)计算值:H5.0860%、C44.4440%、N23.5592%、O26.9108%。元素分析(%)测定值:H5.0829%、C44.4456%、N23.5582%、O26.9133%。元素分析结果与理论值基本一致。采用卡尔·费休水分测定法测定KF为0.44%,所以本发明奈拉滨晶型化合物不含结晶水。所制得的奈拉滨晶型化合物使用Cu‐Kα射线测量得到的X‐射线粉末衍射谱图与实施例1相似;采用美国Perkin‐Elmer公司PEPyrisDiamondTG热分析仪得到的热重分析图与实施例1相似。实施例4(1)将奈拉滨粗品100g加入去离子水中,加热回流,形成65℃下的饱和溶液;(2)加活性炭,过滤,滤液升温至90℃;(3)搅拌速率16rmp下于25min向滤液匀速滴加奈拉滨粗品重量的2.5倍体积的丙酮乙醚(丙酮:乙醚=1:3)混合溶液;(4)滴加完毕后,在转速20rmp下搅拌15min降温至25℃,再在转速10rmp下搅拌15min降温至-5℃,静置析晶19小时,过滤,得奈拉滨晶型化合物湿品;(5)将步骤(4)中得到的奈拉滨晶型化合物湿品在30℃真空烘干,烘干后在惰性气体保护下破除真空冷却至室温,得到高纯度奈拉滨晶型化合物成品94.27g,收率94.27%,光学纯度99.85%。将所制得的奈拉滨晶型化合物用美国Perkin-Elmer公司PE2400Ⅱ元素分析仪进行分析测定:元素分析:元素分析(%)计算值:H5.0860%、C44.4440%、N23.5592%、O26.9108%。元素分析(%)测定值:H5.0839%、C44.4391%、N23.5651%、O26.9119%。元素分析结果与理论值基本一致。采用卡尔·费休水分测定法测定KF为0.41%,所以本发明奈拉滨晶型化合物不含结晶水。所制得的奈拉滨晶型化合物使用Cu‐Kα射线测量得到的X‐射线粉末衍射谱图与实施例1相似;采用美国Perkin‐Elmer公司PEPyrisDiamondTG热分析仪得到的热重分析图与实施例1相似。实施例5(1)将奈拉滨粗品100g加入去离子水中,加热回流,形成75℃下的饱和溶液;(2)加活性炭,过滤,滤液升温至90℃;(3)搅拌速率19rmp下于30min向滤液匀速滴加奈拉滨粗品重量的3.5倍体积的丙酮乙醚(丙酮:乙醚=1:4)混合溶液;(4)滴加完毕后,在转速15rmp下搅拌15min降温至20℃,再在转速15rmp下搅拌15min降温至-5℃,静置析晶16小时,过滤,得奈拉滨晶型化合物湿品;(5)将步骤(4)中得到的奈拉滨晶型化合物湿品在110℃真空烘干,烘干后在惰性气体保护下破除真空冷却至室温,得到高纯度奈拉滨晶型化合物成品94.24g,收率94.24%,光学纯度99.87%。将所制得的奈拉滨晶型化合物用美国Perkin-Elmer公司PE2400Ⅱ元素分析仪进行分析测定:元素分析:元素分析(%)计算值:H5.0860%、C44.4440%、N23.5592%、O26.9108%。元素分析(%)测定值:H5.0889%、C44.4427%、N23.5533%、O26.9151%。元素分析结果与理论值基本一致。采用卡尔·费休水分测定法测定KF为0.39%,所以本发明奈拉滨晶型化合物不含结晶水。所制得的奈拉滨晶型化合物使用Cu‐Kα射线测量得到的X‐射线粉末衍射谱图与实施例1相似;采用美国Perkin‐Elmer公司PEPyrisDiamondTG热分析仪得到的热重分析图与实施例1相似。实验例:本发明还提供了如下实验例,以对本发明产品进一步说明。实验例1、本实验例对实施例1‐5所制备的奈拉滨晶型化合物中的有关物质进行检测本实验按照中国药典2010版第二部附录ⅧP残留溶剂测定法、附录ⅪⅩF药品杂质分析指导原则进行,其结果见表1。表1各实施例样品有关物质的检测结果样品丙酮乙醚其他有关物质实施例10.02%0.02%0.13%实施例20.02%0.03%0.11%实施例30.03%0.01%0.12%实施例40.02%0.03%0.13%实施例50.02%0.03%0.10%实验例2、本实验例考察了本发明提供的奈拉滨晶型化合物和对照品的最大单杂和总杂照高效液相色谱法(中国药典2005年版二部附录VD)测定。色谱条件与系统适用性实验:以十八烷基硅烷键合硅胶为填充剂;以0.02mol/L磷酸二氢钠的水溶液:甲醇(83:17)为流动相;检测波长:210nm。奈拉滨峰理论塔板数应不低于1500。测定:精密称取奈拉滨适量,用流动相溶解并稀释制成每1ml中含奈拉滨0.125mg的溶液,作为供试品溶液。精密量取上述供试品溶液适量,加流动相制成每1ml中含1.25μg的溶液作为对照溶液;照有关物质项下的色谱条件,取对照溶液20μl注入液相色谱仪,调节检测器灵敏度,使主成份色谱峰的峰高约为满量程的15%,再精密量取供试品溶液与对照溶液各20μl,分别注入液相色谱仪,分别记录色谱峰至主成份峰保留时间的3倍。采用以上检测方法测定了3批样品的有关物质,结果见表2。表2各样品有关物质的检测结果样品最大单杂(%)总杂(%)本发明实施例10.020.17本发明实施例20.030.16本发明实施例30.030.16本发明实施例40.030.18本发明实施例50.030.15CN101348511A实施例190.190.31CN103172687A实施例10.150.27市售0.210.35实验例3、本实验例考察了本发明提供的奈拉滨晶型化合物的流动性本实验例通过测定各实施例样品的休止角来评价样品的流动性,具体方法如下:取样品颗粒,从固定的小漏斗中流入圆形的表面皿中,直到得到最高的圆锥体,量取圆锥体高度H和半径R,按tanα=H/R计算休止角α,结果见表3,休止角越大,流动性越差。表3奈拉滨晶型化合物的流动性样品1样品2样品3样品4样品5样品6样品7样品8α34.7°35.1°35.7°35.4°35.5°47.7°47.1°48.5°其中,样品1是实施例1产品;样品2是实施例2产品;样品3是实施例3产品;样品4是实施例4产品;样品5是实施例5产品;样品6是参照专利CN101348511A实施例19制备的奈拉滨化合物;样品7是参照专利CN103172687A实施例1制备的奈拉滨结晶化合物;样品8是市售奈拉滨原料药。从表3可见,与现有技术的奈拉滨化合物相比,本发明奈拉滨化合物具有优异的流动性,有利于提高分装的准确性,并且与其他成分混合时易于混合均匀。实验例4、本实验例本发明奈拉滨晶型化合物的稳定性试验1、加速试验取实施例1制备的样品三批(批号:201406001、201406002、201406003),于温度40±2℃、相对湿度75±5%的条件下放置6个月,分别于0、1、2、3、6个月末取样测定性状、有关物质、含量,结果见表4。表4加速试验结果(温度40±2℃,相对湿度75±5%)从表4看出,本发明奈拉滨晶型化合物在温度40±2℃、相对湿度75±5%的条件下放置6个月,有关物质含量低,各指标均无明显变化,本品质量稳定性好。其他实施例也经过如上加速试验,试验结果与上表相似。2、长期试验取实施例1制备的样品两批(批号:201406001、201406002),于温度25±2℃、相对湿度60±5%的条件下放置6个月,分别于0、3、6、9、12、18、24个月末取样测定性状、有关物质、含量,结果见表5。表5长期试验结果(温度25±2℃,相对湿度60±5%)从表5看出,本发明奈拉滨晶型化合物在温度25±2℃、相对湿度60±5%的条件下放置24个月稳定,各指标均无明显变化。其他实施例也经过如上长期试验,试验结果与上表相似。实验例5、25℃下本发明奈拉滨晶型化合物与现有技术晶型的在水中的溶解度对比样品包括试验品和对照品:试验品:本发明实施例1‐5所制备的样品;对照品1是参照专利CN101348511A实施例19制备的奈拉滨化合物;对照品2是参照专利CN103172687A实施例1制备的奈拉滨结晶化合物;对照品3是市售奈拉滨原料药。测定方法:将过量的试验品和对照品置于50ml锥形瓶中,加入30ml蒸馏水,25℃恒温搅拌72小时,取样5ml。样品经0.45μm微孔滤膜过滤,弃去初滤液,取续滤液20μL测定药物含量即为水中溶解度(mg/ml)。结果见表6:表6奈拉滨晶型化合物与现有技术晶型在水中的溶解度(mg/ml)对比样品溶解度实施例128.25实施例228.17实施例328.11实施例428.06实施例528.15对照品18.7对照品219.1对照品38.8从表6可以看出,25℃下,本发明奈拉滨晶型化合物的在水中的溶解度与现有技术相比,有显著提高。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种抗肿瘤药物奈拉滨化合物的制造方法与工艺
- 四苯基卟啉连二胺芳基钌化合物及其制备方法和用途与制造工艺
- 一种具抗肿瘤活性的不对称双1‑取代5‑N杂靛红席夫碱类化合物的合成方法与制造工艺
- 一种具抗肿瘤活性的不对称双7‑N杂靛红席夫碱类化合物的合成方法与制造工艺
- 一种具抗肿瘤活性的不对称双取代靛红席夫碱类化合物及其合成方法与制造工艺
- 一种抗肿瘤药物奈拉滨组合物的制造方法与工艺
- 含Micranthanone A用于抗肿瘤的药物的制造方法与工艺
- 一种小分子靶向药物脂质体制剂的制备方法与制造工艺
- 一种5?溴?1H?吡咯并[2,3?b]吡啶的合成方法
- 一种作用于EGFR敏感突变激酶EGFR<sup>L858R</sup>,EGFR<sup>(d746-750)</sup>的6-取代氨基嘌呤类化合物及 ...的制作方法