用于制备取代的5‑氟‑1H‑吡唑并吡啶类化合物的方法与流程
本申请涉及用于制备新型的式(VI)的取代的5-氟-1H-吡唑并吡啶类化合物的新颖和有效的方法,
该化合物作为中间体用于制备药物和制备用于治疗和/或预防心血管疾病的药物。
更具体地,式(VI)的5-氟-1H-吡唑并吡啶类化合物适用于制备式(I)的化合物,
该化合物用于制备药物和制备用于治疗和/或预防心血管疾病的药物。
式⑴的化合物起到可溶性鸟苷酸环化酶刺激剂的作用,并可用作预防和/或治疗心血管疾病的药剂,例如用于治疗高血压和心脏衰竭、稳定型和不稳定型心绞痛、外周和心血管病症、心律不齐、用于治疗血栓栓塞性病症和缺血例如心肌梗塞、中风、短暂性和缺血性发作、外周灌注紊乱、预防再狭窄例如血栓疗法、经皮腔内血管成形术(PTA)、经皮腔内冠状动脉成形术(PTCA)、分流术,和用于治疗动脉硬化、哮喘病症和泌尿生殖系统疾病,例如前列腺肥大、勃起功能障碍、女性性功能障碍、骨质疏松症、青光眼、肺动脉高压、胃轻瘫、硬皮病和尿失禁。
式⑴化合物可以以不同的晶型和溶剂合物存在。式⑴化合物以具有熔点257℃ (多晶型物(Modifikation)I)、253℃ (多晶型物II)、247℃ (多晶型物III)、246℃ (多晶型物IV)、234℃ (多晶型物V)的五种多晶型物,以及以二甲基甲酰胺/水-溶剂合物(DMF含量13.6%,水含量0.9%)、二-二甲基亚砜-溶剂合物(化学计量值: 26.8% DMSO)、三乙酸-溶剂合物(29.7% 乙酸酯)、单水合物(4.1% 水)和二水合物(7.8% 水)存在。现有技术WO 2011/147809在实施例1的物质中描述了式(I)的化合物。
以多晶型物(I)存在的式(I)的化合物的结晶多晶型物以稳定性引人注目,特别是其甚至在微粒化过程中保持稳定因此不会发生转化和重结晶。
式(I)的化合物的二-二甲基亚砜-溶剂合物比现有技术中的物质具有明显更好的可过滤性的优点。此外,通过式(I)的化合物的二-二甲基亚砜-溶剂合物的制备方法可以获得非常高纯度的式(I)的化合物。
WO 03/095451、WO 2011/064156和WO 2011/064171中公开了在吡啶环上未取代的吡唑并吡啶类化合物的合成。在这些公开文件中,双环环体系通过苯基苄基肼与氰基丙酮酸乙酯的反应建立。这种合成方法不适用于形成5-氟-1H-吡唑并吡啶类化合物。
WO 2009/018415描述了5-氟-1H-吡唑并[3,4-b]吡啶-3-胺E的合成。通过将烟酸A选择性脱氯获得化合物B,随后转化为酰胺C,将其还原为腈并最后用肼水合物环化形成5-氟-1H-吡唑并[3,4-b]吡啶核。以下示意图1说明了该合成。
示意图1:
。
这种方法的缺点是,为了获得所希望的式(VI)的5-氟-1H-吡唑并吡啶类化合物,从5-氟-1H-吡唑并[3,4-b]吡啶 E开始,需要其它步骤例如重氮化反应以及转化为碘化合物,接着通过用苄基衍生物烷基化以及随后功能化以引入氰基基团。示意图2中通过示例的方式说明了该方法。
示意图2:
另一个缺点是,重氮化在无水条件下实施并且必须分离重氮盐,在将其转换为工业规模时,大量的安全性预防措施是必需的,因此会导致高的生产成本。
另一个缺点是,用苄基衍生物烷基化非选择性地进行,并且在复杂的纯化和分离异构体后仅得到低产率的产物。
再一个缺点是,在氰化过程中,必须处理有毒的氰化铜,在制备中和在母液以及水相的处理中都必须采取另外的安全预防措施,因此导致高的生产成本。
再另一个缺点是,根据示意图1中描述的方法制备式(VI)的5-氟-1H-吡唑并吡啶类化合物,必须制备和纯化七个中间体并仅得到小的总产率。
本发明的目的是提供用于制备式(VI)的5-氟-1H-吡唑并吡啶类化合物的高产率有效方法,
式(VI)的5-氟-1H-吡唑并吡啶类化合物是用于制备式(I)的化合物
及其N-氧化物、盐类、溶剂合物、N-氧化物的盐类和N-氧化物的溶剂合物和盐类的溶剂合物的高产率有效方法的关键组分。
此目的根据本发明的如下内容实现。以下示意图3通过示例的方式说明每个反应步骤。
示意图3:
[a): LiCl, MeSO3H, EtOH; b)甲酰胺, NaOMe/MeOH, EtOH; c) POCl3, CH3CN, 环丁砜; d) 1. NaOMe/MeOH, 2. NH4Cl/EtOH; e) DMF, NEt3, 苯基偶氮基丙二腈; f) Pd/C, H2, DMF; g) iPrOH, 氯甲酸甲酯, NEt3]。
通过(WO 03/004503 (实施例 IIIb)和WO 03/095451 (实施例 2A)),已知步骤a)已经用于未取代的吡唑并吡啶类化合物:
[aa): CF3SO3H, 回流三天, 层析, 产率49.9%]。
与现有技术相比(WO 03/004503, 实施例 IIIb 和WO 03/095451, 实施例 2A),IV的制备产生更高的产率。
另一个优点是,不使用腐蚀性的三氟乙酸,而使用便宜得多的乙醇作为溶剂。
另一个优点是,与现有技术相比,反应时间相当短。
另一个优点是,IV的制备具有高选择性,并且产物以高纯度形成而没有大量的副产物形成,以及不需要复杂的纯化程序。
另一个优点是,以结晶得到高产率和高纯度的IV。
通过WO 03/095451、WO 2011/064156和WO 2011/064171,已知步骤d)–g)已经用于未取代的吡唑并吡啶类化合物,并且可以类似地使用。
特别地,根据本发明用于制备式(VI)的化合物的方法
包括5-氨基吡唑衍生物 (IIa)
其中
T1 是(C1-C4)-烷基,
在适当的酸的存在下与醛(III)环化
其中,R1和R2各自独立地是甲基、乙基、异丙基、苯基或与它们所键接至的氮原子一起,是
以获得式(IVa)的酯,
其中T1如上所定义,
其后续与氨或甲酰胺反应以获得式(V)的酰胺
以及随后脱水以获得腈(VI)。
本发明还提供式(VI)的化合物
用于制备(I)的化合物
及其N-氧化物、盐类、溶剂合物、N-氧化物的盐类和N-氧化物的溶剂合物和盐类的溶剂合物的用途。
本发明还提供式(III)的化合物
其中 R1和R2 各自独立地是甲基、乙基、异丙基、苯基或,与它们所键接至的氮原子一起,是
用于制备式(I)的化合物
及其N-氧化物、盐类、溶剂合物、N-氧化物的盐类和N-氧化物的溶剂合物和盐类的溶剂合物的用途。
本发明进一步提供式(VI)的化合物用于制备如上所述的式(I)的化合物的用途,其中,将式(VI)的化合物转化为式(VII)的化合物,
式(VII)的化合物随后在惰性溶剂中在合适的碱的存在下与式(VIIIa)的化合物反应
以获得式(VIII)的化合物,
然后,式(VIII)的化合物在惰性溶剂中在合适的还原剂的存在下被还原,以获得化合物(IX),
然后化合物 (IX)在合适的碱的存在下,在有或没有溶剂的情况下,与氯甲酸甲酯或与二碳酸二甲酯反应,以获得式(I)的化合物,
得到的式(I)的化合物任选地用合适的(i)溶剂和/或(ii)酸或碱任选地转化为其溶剂合物、盐类和/或盐类的溶剂合物。
(VI) → (VII)的转化通过本领域技术人员所知的方法以两步法来实施,首先在甲醇中在0℃至+40℃下与甲醇钠形成亚氨酯,然后在+50至+150℃下,在乙酸或醇中与氨同等物,例如氨或氯化铵进行亲核加成以形成脒(VII)。
用于(VI) → (VII)转化的合适的醇是醇类,例如甲醇、乙醇、正丙醇、异丙醇、正丁醇或叔丁醇。
用于(VII) + (VIIIa) → (VIII)方法步骤的惰性溶剂是醇类,例如甲醇、乙醇、正丙醇、异丙醇、正丁醇或叔丁醇,醚类,例如乙醚、二氧六环、四氢呋喃、乙二醇二甲醚或二乙二醇二甲醚,烃类,例如苯、二甲苯、甲苯、己烷、环己烷或石油馏份,或其它溶剂,例如二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、环丁砜、N,N'-二甲基亚丙基脲(DMPU)、N-甲基吡咯烷酮(NMP)、吡啶、乙腈或水。同样可以使用所述溶剂的混合物。优选使用的是DMF和环丁砜。
用于(VII) + (VIIIa) → (VIII)方法步骤的合适的碱为碱金属氢氧化物,例如氢氧化锂、氢氧化钠或氢氧化钾,碱金属碳酸盐例如碳酸锂、碳酸钠、碳酸钾或碳酸铯,碱金属碳酸氢盐,例如碳酸氢钠或碳酸氢钾,碱金属醇盐,例如甲醇钠或甲醇钾、乙醇钠或乙醇钾或叔丁醇钾,或有机胺类例如三乙胺、二异丙基乙基胺、吡啶、1,8-二氮杂双环[5.4.0]十一-7-烯(DBU)或1,5-二氮杂双环[4.3.0]壬-5-烯(DBN)。优选使用的是三乙胺。
反应(VII) + (VIIIa) → (VIII)一般在+20℃至+150℃,优选在 +80℃至+120℃下,任选在微波中实施。该转化可在标准、升高或降低的压力(例如0.5-5巴)下实施。通常使用标准压力。
式(VIIIa)的化合物可以类似于文献L. F. Cavalieri, J. F. Tanker, A. Bendich, J. Am. Chem. Soc., 1949, 71, 533来制备。
还原反应(VIII) → (IX)在合适的催化剂的存在下,在惰性溶剂中,在+20℃至+100℃的温度范围内,在氢气压力(例如1-100巴)下实施。优选使用40℃ 至 80℃的温度范围和5-70巴的氢气压力范围。
用于还原反应(VIII) → (IX)的惰性溶剂为,例如,醇类例如甲醇、乙醇、正丙醇、异丙醇、正丁醇或叔丁醇,醚类,例如乙醚、二氧六环、四氢呋喃、乙二醇二甲醚或二乙二醇二甲醚,或其它溶剂,例如二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、N,N'-二甲基亚丙基脲(DMPU)、N-甲基吡咯烷酮(NMP)、吡啶、乙腈或水。同样可以使用所述溶剂的混合物。优选使用的是DMF和吡啶。
用于转化(VIII) → (IX)的合适的催化剂为,例如,活性炭载钯、炭载铂、氢氧化钯或Raney镍。
还原反应(VIII) → (IX)可供选择地用金属或金属盐,例如铁、锌或氯化锡(II),在合适的酸,例如盐酸、硫酸、磷酸或乙酸中在+20℃至+140℃的温度范围内进行。
用于方法步骤(IX) → (I)的惰性溶剂是,例如,醇类例如甲醇、乙醇、正丙醇、异丙醇、正丁醇或叔丁醇,醚类,例如乙醚、二异丙基醚、二氧六环、四氢呋喃、乙二醇二甲醚或二乙二醇二甲醚,卤代烃类,例如二氯甲烷、三氯甲烷、四氯化碳、三氯乙烷或氯苯,烃类,例如苯、二甲苯、甲苯、己烷、环己烷或石油馏份,或其它溶剂,例如二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、N,N'-二甲基亚丙基脲(DMPU)、N-甲基吡咯烷酮(NMP)、乙腈、乙酸乙酯或水。同样可以使用所述溶剂的混合物。优选使用的是异丙醇和四氢呋喃以及异丙醇和四氢呋喃的混合物。
用于方法步骤(IX) → (I)的合适的碱为碱金属氢化物,例如氢化钠,碱金属氢氧化物,例如氢氧化锂、氢氧化钠或氢氧化钾,碱金属碳酸盐例如碳酸锂、碳酸钠、碳酸钾或碳酸铯,碱金属碳酸氢盐,例如碳酸氢钠或碳酸氢钾,碱金属醇盐,例如甲醇钠或甲醇钾、乙醇钠或乙醇钾或叔丁醇钾,或有机胺类例如三乙胺、二异丙基乙基胺、吡啶、4-二甲基氨基吡啶、1,8-二氮杂双环[5.4.0]十一-7-烯(DBU)或1,5-二氮杂双环[4.3.0]壬-5-烯(DBN)。优选使用的是三乙胺。
反应(IX) → (I)通常在-10℃至+70℃,优选0℃至+50℃的温度范围内实施。该转化可以在标准、升高或降低的压力(例如0.5-5巴)下实施。通常使用标准压力。
式(IIa)的化合物从文献中是已知的,并且可以类似于WO 00/06569中实施例20A来制备。
式(III)的化合物从文献H. Yamanaka, S. Yamashita和T. Ishihara, Synlett 353-354 (1993)中是已知的。示意图4中说明了其中公开的合成。
示意图4:
[k) 3当量 二甲基苄基胺, 130-140℃; l) 10当量 CH3I, 回流; m) 1M NaOH, 20℃; n) DMSO-H2O (1:1), 吗啉, 40℃, 3h]。
这种方法的缺点是,根据H. Yamanaka, M. Kuwabara, M. Okudo, K. Fukunishi和M. Nomura, Nippon Kagaku Kaishi (10) 1988-1994 (1985),在(XVIb)的制备中,仅达到66%的产率,并且在此方法中,得到非常大量(每kg的(XVIb)得到2.79 kg)的副产物(硝基苯磺酸二甲基二苄基酯),所述副产物必须移除和处理掉。
此方法的另一缺点是,根据Yamanaka, H. Ganbayashi, M. Kuwabara, K. Fukunishi和M. Nomura, Nippon Kagaku Kaishi (7) 1036-1043 (1988),从(XVIb)开始,烷基化作用要求10当量的致癌的烷基化试剂碘甲烷。
此方法的另一缺点是,根据H. Yamanaka, S. Yamashita和T. Ishihara, Synlett 353-354 (1993),O与吗啉的反应不仅形成所希望的产物(IIIb),而且还形成11%的副产物(IIIa),其需要复杂的纯化,结果是,用于制备(IIIb)的总合成仅得到低的总产率,并导致高的生产成本。
然而,其中所述的合成不适合在工业规模中用于制备式(III)的醛类,所以已经开发出一种新颖和有效的合成,所述合成在示意图5中通过示例的方式来说明。
示意图5:
[o)无溶剂; p) 二氯甲烷或无溶剂, 吗啉; q) 无溶剂, 甲磺酸甲酯; r) NaOH, 水; s) 吗啉/三乙胺]。
式(XIII)的化合物由文献Markovskii, L. N.; Kolesnik, N. P.; Shermolovich, Yu. G Zhurnal Obshchei Khimii (1980), 50(4), 826-829是已知的。其中所公开的合成在示意图6中说明。
示意图6:
然而,其中所述的合成,主要出于低产率的原因,不适合在工业规模上制备式(III)的醛类。
本发明还提供用于制备式(III)的化合物的方法,
其中R1 和R2各自独立地是甲基、乙基、异丙基、苯基或,与它们所键接至的氮原子一起,是
其中式(X)的三氟甲磺酸酐是在无溶剂的条件下与式(XI) 的2,2,3,3-四氟-1-丙醇反应并将得到的式(XII)的2,2,3,3-四氟丙基三氟甲磺酸酯与式(XIIa)的化合物反应,
其中R1和R2各自如上所定义,
以获得式(XIIIa)的化合物,
其中R1和R2各自如上所定义,
和与甲磺酸甲酯反应以获得式(XIVa)的化合物,
其中R1和R2各自如上所定义,
和与氢氧化钠反应以获得式(XVa)的化合物,
其中R1和R2各自如上所定义,
和最后在碱性条件下转化以获得式(III)的化合物。
本发明进一步优选提供用于制备式(IIIa)的化合物的方法,
其中式(X)的三氟甲磺酸酐在无溶剂的条件下与式(XI)的2,2,3,3-四氟-1-丙醇反应,并将得到的式(XII) 2,2,3,3-四氟丙基三氟甲磺酸酯与吗啉反应以获得式(XIII)的化合物,
以及与甲磺酸甲酯反应以获得式(XIV)的化合物,
以及与氢氧化钠反应以获得式(XV)的化合物,
以及最后加入吗啉以获得式(III)的化合物。
此新合成优于现有技术的优点是,中间体(XII)和当前未知的中间体 (XIV)和(XV)不需要分离,这大大降低了合成的工业复杂性。
用新的合成方法获得的式(III)的醛类的产率比现有技术要高很多。
用于(XIVa)至(XVa)方法步骤的本发明的上下文中的“碱性条件”是指在该反应中所形成的酸被辅助碱类,例如氢氧化钠、氢氧化钾、碳酸钾、碳酸钠或三乙胺所清除,以形成对应的盐类。
与现有技术相比,(XIII)的制备以明显更高的产率进行。有利的是,(XII)的制备不需要溶剂,而且在后续阶段中不需要进一步纯化地使用中间体XII从而获得(XIII)。
此方法的另一优点是,在(XIII)的制备中没有形成明显的废物。还有利的是,三氟甲磺酸和吗啉可从所形成的三氟甲磺酸吗啉鎓中回收。
与现有技术相比,(XIV)的制备仅需要一当量的烷基化试剂。此反应在无溶剂下实施并且实质上定量实施,其获得了高的时空产率。
此方法的另一优点是,不用分离产物(XIV),将(XIV)溶解于水中,并使该溶液与氢氧化钠溶液反应以获得(XV)。
此方法的另一优点是,产物(XV)也不用分离,其水溶液与吗啉的反应得到高产率的单一产物(IIIa)。
此方法的另一优点是,(IIIa) 通过结晶以高的总产率和纯度获得。
化合物(IIa)的5-氨基吡唑衍生物与化合物 (III)的醛获得式(IV)的化合物的环化反应在惰性溶剂中,在+10℃至+200℃,优选在+20℃至+100℃的温度范围内,在标准压力下,在例如2-50小时,优选2-20小时内,任选地在酸和任选的碱金属盐的存在下实施。
酸是,例如,盐酸、三氟乙酸和甲磺酸。优选使用甲磺酸和盐酸。
碱金属盐类是氯化钠或氯化锂。优选的碱金属盐是氯化锂。
惰性溶剂为,例如,醇类例如甲醇、乙醇、正丙醇、异丙醇、正丁醇,醚类,例如乙醚、二氧六环、四氢呋喃、乙二醇二甲醚或二乙二醇二甲醚,烃类,例如苯、甲苯、二甲苯、己烷、环己烷或石油馏份,或其它溶剂,乙腈或N,N'-二甲基甲酰胺,或溶剂的混合物。优选使用的是乙醇、二乙二醇二甲醚或二氧六环。
酰胺(IVa) → (V)的优选形成通过在惰性溶剂中,在碱的存在下,在0℃至+150℃, 优选+20℃至+130℃的温度范围内,在标准压力或升高的压力下,在2-24小时内与甲酰胺反应来实施。
惰性溶剂是,例如醇类如甲醇、乙醇、正丙醇或异丙醇。优选使用乙醇。
用于优选方法步骤(IVa) → (V)的合适的碱是碱金属碳酸盐例如碳酸锂、碳酸钠、碳酸钾或碳酸铯,碱金属碳酸氢盐,例如碳酸氢钠或碳酸氢钾,碱金属醇盐,例如甲醇钠或甲醇钾、乙醇钠或乙醇钾或叔丁醇钾,或有机胺类例如三乙胺、二异丙基乙基胺、吡啶、1,8-二氮杂双环[5.4.0]十一-7-烯(DBU)或1,5-二氮杂双环[4.3.0]壬-5-烯(DBN)。优选使用的是甲醇钠和乙醇钠。
酰胺(IVa) → (V)的形成可供选择地在0℃至+50℃,优选+20℃至+30℃的温度范围内,在标准压力或升高的压力下,在24-72小时内与氨反应来实施。
惰性溶剂是,例如,醇类例如甲醇、乙醇、正丙醇或异丙醇。优选使用氨在甲醇中浓度为5N-7N的溶液。
酰胺(V)脱水以形成腈(VI)的反应在惰性溶剂中,任选在合适的碱的存在下,在0℃至+150℃,优选+50℃至+110℃的温度范围内,在1-12小时内,与合适的脱水剂例如氧氯化磷、三氟乙酸酐、乙酸酐或三氟甲磺酸酐反应来实施。
优选使用的是氧氯化磷。
惰性溶剂为醚类,例如乙醚、二氧六环、四氢呋喃(THF)、乙二醇二甲醚或二乙二醇二甲醚,烃类,例如苯、甲苯、二甲苯、己烷、环己烷或石油馏份,或其它溶剂,吡啶、环丁砜、乙腈或N,N'-二甲基甲酰胺,或溶剂的混合物。优选使用的是环丁砜和乙腈。
合适的碱是,例如,有机胺类例如三乙胺、二异丙基乙基胺、吡啶、1,8-二氮杂双环[5.4.0]十一-7-烯(DBU)或1,5-二氮杂双环[4.3.0]壬-5-烯(DBN)。优选使用的是吡啶。
在根据本发明的方法的上下文中所描述的化合物还可以是其盐类、溶剂合物或盐类的溶剂合物的形式。
在根据本发明的方法的上下文中所描述的化合物,取决于结构,还可以是其互变异构体的形式。
在本发明的上下文中优选的盐类是在根据本发明的方法中所使用和制备的化合物的生理上可接受的盐类。
在根据本发明的方法中所使用和制备的化合物的生理上可接受的盐类包括无机酸、羧酸和磺酸的酸加成盐类,例如盐酸、氢溴酸、硫酸、磷酸、甲磺酸、乙磺酸、甲苯磺酸、苯磺酸、萘二磺酸、乙酸、丙酸、乳酸、酒石酸、苹果酸、柠檬酸、富马酸、马来酸和苯甲酸的盐类。
在根据本发明的方法中所使用和制备的化合物的生理上可接受的盐类还包括惯用碱的盐类,示例和优选的是,碱金属盐类(例如钠盐和钾盐)、碱土金属盐类(例如钙盐和镁盐)以及衍生自氨或具有 1至16个碳原子的有机胺的铵盐类,示例和优选的是,乙胺、二乙 胺、三乙胺、乙基二异丙基胺、单乙醇胺、二乙醇胺、三乙醇胺、二环己胺、二甲基氨基乙醇、普鲁卡因、二苄基胺、N-甲基吗啉、二氢松香胺、精氨酸、赖氨酸、乙二胺和甲基哌啶。
在本发明的上下文中,溶剂合物是指在根据本发明的方法中所使用和制备的化合物的那些形式,其以固态或液态通过与溶剂分子配位形成配合物。水合物是溶剂合物的特定形式,其中与水进行配位。
在本发明的上下文中,除非另有说明,否则取代基各自如下所定义::
烷基在本发明的上下文中是指具有1至4个碳原子的直链或支化烷基。优选的实例包括:甲基、乙基、正丙基、异丙基、 正丁基、异丁基、仲丁基和叔丁基。
下面通过非限制性的优选实施例和比较实施例详细说明本发明。除非另有说明,否则所有给出的量都是指重量百分比。
本发明提供用于制备式(VI)的化合物的方法,
其特征在于式(V)的化合物
通过式(IVa)的酯与甲酰胺的反应来制备,
其中,
T1是(C1-C4)-烷基。
本发明进一步提供如上所述的方法,其特征在于,式(IVa)的酯通过5-氨基吡唑衍生物 (IIa)
其中
T1是(C1-C4)-烷基,
在酸和碱金属盐的存在下与式(III)的醛
其中 R1和R2各自独立地是甲基、乙基、异丙基、苯基或,与它们所键接至的氮原子一起,是
进行环化反应来制备。
本发明进一步提供如上所述的方法,其特征在于,在环化反应中所使用的醛是式(IIIa)的化合物
。
本发明进一步提供用于制备式(III)的醛类的方法,
其中R1和R2各自独立地是甲基、乙基、异丙基、苯基或,与它们所键接至的氮原子一起,是
其特征在于,三氟甲磺酸酐在没有溶剂的条件下与2,2,3,3-四氟-1-丙醇反应并且得到的2,2,3,3-四氟丙基三氟甲磺酸酯与式(XIIa)的化合物反应,
其中R1和R2 各自如上所定义,以获得式(XIIIa)的化合物,
其中R1和R2 各自如上所定义,
和与甲磺酸甲酯反应以获得式(XIVa)的化合物,
其中 R1和R2 各自如上所定义,
和与氢氧化钠反应以获得式(XVa)的化合物,
其中R1和R2 各自如上所定义,
并最终在碱性条件下转化以获得式(III)的化合物。
本发明进一步提供用于制备式(IIIa)的化合物的方法,
其中式(X)的三氟甲磺酸酐在没有溶剂的条件下与式(XI) 的2,2,3,3-四氟-1-丙醇反应,并且得到的式(XII)的2,2,3,3-四氟丙基三氟甲磺酸酯与吗啉反应以获得式(XIII)的化合物,
和与甲磺酸甲酯反应以获得式(XIV)的化合物,
和与氢氧化钠反应以获得式(XV)的化合物,
和最后通过加入吗啉反应以获得式(IIIa)的化合物。
本发明进一步提供用于制备式(I)的化合物的方法,
其特征在于,使用式(VI)的化合物,
式(VI)的化合物的特征在于,它们通过如上详述的方法制备,并任选地用合适的(i)溶剂和/或(ii)酸类或碱类任选地将得到的式(I)的化合物转化为其溶剂合物、盐类和/或盐类的溶剂合物。
本发明进一步提供用于制备式(I)的化合物的方法,其特征在于,使用式(VI)的化合物,
式(VI)的化合物的特征在于,它们通过如上详述的方法制备,并任选地用合适的(i)溶剂和/或(ii)酸类或碱类任选地将得到的式(I)的化合物转化为其溶剂合物、盐类和/或盐类的溶剂合物。
本发明进一步提供用于制备式(I)的化合物的方法,其特征在于,使用式(VI)的化合物,
式(VI)的化合物的特征在于,它们通过如上详述的方法制备,并任选地用合适的(i)溶剂和/或(ii)酸类或碱类任选地将得到的式(I)的化合物转化为其溶剂合物、盐类和/或盐类的溶剂合物。
本发明进一步提供用于制备式(I)的化合物的方法,其特征在于,使用式(VI)的化合物,式(VI)的化合物通过如上详述的方法制备,其中将式(VI)的化合物转化为式(VII)的化合物,
随后将式(VII)的化合物在惰性溶剂中,在合适的碱的存在下,与式(VIIIa)的化合物反应
以获得式(VIII)的化合物,
然后在惰性溶剂中,在合适的还原剂的存在下,还原式(VIII)的化合物以获得化合物(IX),
然后式(IX)的化合物在有或没有溶剂的条件下,在合适的碱的存在下,与氯甲酸甲酯或二碳酸二甲酯反应得到式(I)的化合物,并任选地用合适的(i)溶剂和/或(ii)酸类或碱类任选地将所得到的式(I)的化合物转化为其溶剂合物、盐类和/或盐类的溶剂合物。
本发明进一步提供以多晶型物I的结晶形式存在的式(I)的化合物,
其特征在于,该化合物的X射线衍射图在5.9、6.9、22.7处具有2θ角的最大波峰。
本发明进一步提供如上所描述的以多晶型物(I)存在的式(I)的化合物,其特征在于,该化合物的X射线衍射图在5.9、6.9、16.2、16.5、24.1、22.7、24.7处具有2θ角的最大波峰。
本发明进一步提供以多晶型物I的结晶形式存在的式(I)的化合物,
其特征在于该化合物的IR光谱在1707、1633、1475 cm-1处具有最大谱带。
本发明进一步提供如上所描述的以多晶型物(I)存在的式(I)的化合物,其特征在于,该化合物的IR光谱在1707、1633、1566、1475、1255、1223 cm-1处具有最大谱带。
本发明进一步提供用于制备以多晶型物I的结晶形式存在的式(I)的化合物的方法,其特征在于,以一种或多种多晶型物存在或作为溶剂合物存在于惰性溶剂中的式(I)的化合物在20℃-120℃的温度下搅拌,并且分离结晶多晶型物I形式的式(I)的化合物。
用于制备以多晶型物I的结晶形式存在的式(I)化合物的方法的优选溶剂为乙酸乙酯/乙醇/水的混合物、异丙醇、异丙醇/水的混合物、甲醇、甲醇/水的混合物、乙腈、丙酮、四氢呋喃和甲基叔丁基醚。
用于制备以多晶型物I的结晶形式存在的式(I)化合物的方法的优选温度范围是20℃至90℃。
本发明进一步提供用于治疗疾病的如上所述的以多晶型物(I)存在的式(I)的化合物。
本发明进一步提供药物,所述药物包括如上所述以多晶型物(I)存在的式(I)的化合物和无更大比例的另一形式的如上所述以多晶型物(I)存在的式(I)的化合物。本发明进一步提供药物,基于如上所述以多晶型物(I)所含的中式(I)的化合物的总重量,所述药物包括大于90重量%的如上所述以多晶型物(I)存在的式(I)的化合物。
本发明进一步提供如上所述以多晶型物(I)存在的式(I)的化合物用于制造治疗心血管疾病的药物的用途。
本发明进一步提供通过施用有效量的如上所述以多晶型物(I)存在的式(I)的化合物来治疗心血管疾病的方法。
本发明进一步提供作为二-二甲基亚砜-溶剂合物的式(I)化合物,
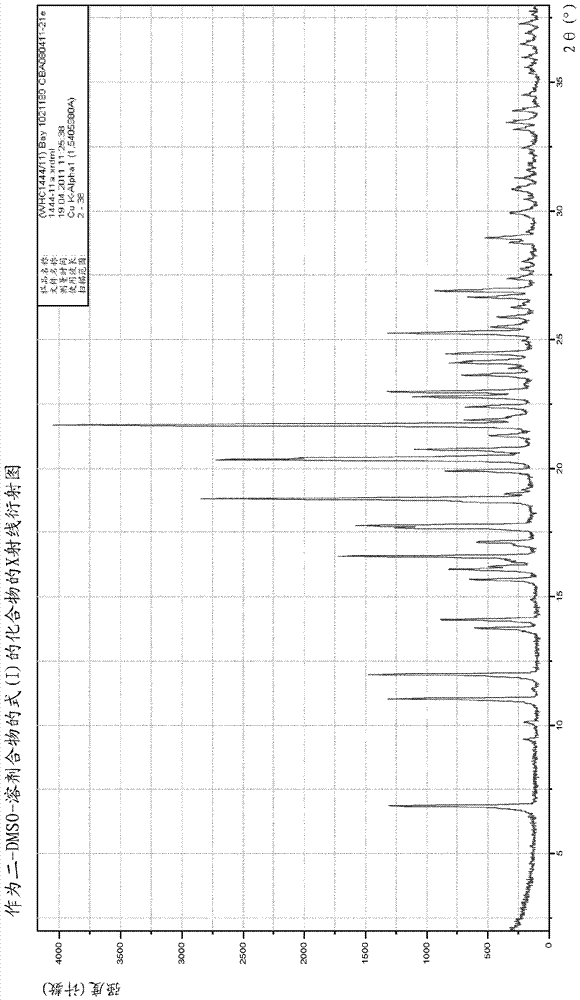
其特征在于,该化合物的X射线衍射图在18.8、20.3、21.7处具有2θ角的最大波峰。
本发明进一步提供作为二-二甲基亚砜-溶剂合物的式(I)化合物,其特征在于,该化合物的X射线衍射图在12.0、16.6、17.8、18.8、20.3、21.7处具有2θ角的最大波峰。
本发明进一步提供作为二-二甲基亚砜-溶剂合物的式(I)化合物,
其特征在于该化合物的IR光谱在1720、1628、1481 cm-1处具有最大谱带。
本发明进一步提供作为二-二甲基亚砜-溶剂合物的式(I)化合物,其特征在于,该化合物的IR光谱在1720、1628、1481、1234、1041、1017 cm-1处具有最大谱带。
本发明进一步提供用于制备结晶形式的作为二-二甲基亚砜-溶剂合物的式(I)化合物的方法,其特征在于,以一种或多种多晶型物存在或作为溶剂合物存在于二甲基亚砜或二甲基亚砜和惰性溶剂(例如乙酸乙酯)的混合物中的式(I)的化合物在20℃-120℃的温度下搅拌,并分离所述二-二甲基亚砜-溶剂合物。优选使用的温度范围是20℃-90℃。
本发明进一步提供式(XIV)的化合物
及其盐类、溶剂合物和盐类的溶剂合物。
本发明进一步提供式(XV)的化合物
及其盐类、溶剂合物和盐类的溶剂合物。
A.实施例
缩写:
Ac 乙酰基
CI 化学电离 (在MS中)
DCI 直接化学电离 (在MS中)
DMF 二甲基甲酰胺
DMSO 二甲基亚砜
d.Th. 理论值的(产率)
eq. 当量
ESI 电喷射电离(在MS中)
Et 乙基
GC/MS 气相色谱-质谱联动
sat. 饱和的
h 小时
HPLC 高压高效液相色谱
HV 高真空
conc. 浓缩的
LC/MS 液相色谱质谱联动
Me 甲基
min 分钟
MS 质谱法
NMR 核磁共振波谱法
rac 外消旋的/消旋体
Rf 保留因子 (在硅胶柱上的薄层色谱中)
RT 室温
Rt 保留时间 (在HPLC中)
SFC 超临界流体色谱法
THF 四氢呋喃
UV 紫外光谱法
v/v 体积/体积比(溶液的)。
所有X射线衍射数据用以下采集参数获得:
衍射仪系统 PANalyticalXPERT-PRO
扫描轴 Gonio
阳极材料 Cu
K-α1 [Å] 1.54060
Kα2 [Å] 1.54443
K-A2 / K-A1比例 0.50000
扫描模式: 透射
扫描类型: 2θ:Ω
2θ值: ±0.2°。
所有红外光谱数据用以下采集参数获得:
光谱仪: 带有钻石ATR单元的Perkin Elmer Spectrum One
参数: 32 次扫描
分辨率: 2 cm-1。
实施例1
2,2,3,3-四氟丙基三氟甲磺酸酯
方法A:
将252.5 g (0.895 mol)三氟甲磺酸酐加热至40℃以及在此温度下计量加入130.0 g (0.984 mol)2,2,3,3-四氟-1-丙醇,同时冷却。计量添加结束后,将反应混合物加热至70-75℃并搅拌2 h。将混合物冷却至20℃并在实施例2的反应中使用该反应溶液而无需进一步纯化。
方法B:
将50.0 g (0.379 mol) 2,2,3,3-四氟-1-丙醇冷却至0℃并在0-4℃下逐滴加入106.8 g (0.379 mol)三氟甲磺酸酐。随后,在25℃下搅拌反应混合物2 h,加热至70-75℃并搅拌2 h。将反应混合物冷却至20℃,并将反应溶液在116-118℃下蒸馏,由此得到85.1 g (理论值的85.1 %) 的标题化合物。
实施例2
4-(2,2,3,3-四氟丙基)吗啉
方法A:
将311.9 g (3.58 mol)吗啉溶解于290 ml的二氯甲烷中,并冷却至-15℃。在-15-0℃下,逐滴加入 371.4 g (最多0.895 mol) 实施例1 的反应溶液,同时冷却,然后将反应混合物在0-5℃ 下搅拌30分钟。将反应混合物加热至40℃并搅拌4.5 h。冷却至20℃后,加入320 ml的水并分离各相。将有机相洗涤三次,每次用190 ml的水,并在30℃/30 毫巴下在旋转蒸发仪上浓缩。残余物 (160.7 g)在67-68℃/18 毫巴下蒸馏。由此获得151.7 g (理论值的84.3 %)的标题化合物。
方法B:
将158.5 g (1.82 mol) 的吗啉冷却至5℃。在5-10℃下,逐滴加入189.5 g (最多0.455 mol)实施例1的反应溶液,同时冷却,然后将混合物在5-10℃下搅拌30分钟。将反应混合物加热至40℃并搅拌1 h 。冷却至20℃后,加入160 ml 的水和160 ml 的甲苯并分离各相。有机相用160 ml 的水洗涤并在50℃/50 毫巴下在旋转蒸发仪上蒸馏。在67-68℃/18 毫巴下蒸馏残余物(81.0 g) 由此获得77.0 g (理论值的84.1 %) 的标题化合物。
实施例3
4-甲基-4-(2,2,3,3-四氟丙基)吗啉-4-鎓甲磺酸盐
方法A:
将143.7 g (1.31 mol)的甲磺酸甲酯加热至135℃,并在此温度下逐滴加入250.0 g (1.243 mol) 的实施例2的化合物。随后,在100℃下搅拌该混合物22 h.。将反应混合物冷却至85℃,并加入375 ml 的异丙醇。冷却至0-5℃后,另外搅拌该混合物30分钟,并通过抽气将产物滤出。产物用异丙醇洗涤三次,每次用异丙醇125 ml,在真空干燥箱中在45℃以及温和的氮气流下干燥。由此获得 336.8 g (理论值的87.1% ) 的标题化合物。
方法B:
将20.0 g (181.3 mmol)的甲磺酸甲酯加热至135℃,并在此温度下逐滴加入35.1g (172.7 mmol) 的实施例2的化合物。该混合物在135℃下搅拌3 h,然后加入40 ml的水。冷却至50℃后,在后续阶段(参见实施例4)中使用标题化合物的水溶液。
实施例4
4-甲基-4-[2,3,3-三氟丙-1-烯-1-基]吗啉-4-鎓甲磺酸盐
将16.9 g (189.9 mmol)45%的氢氧化钠溶液在50-55℃下计量加入实施例3方法B化合物的水溶液(最多172.7 mmol)中,并在50℃下搅拌该混合物1 h。将反应混合物冷却至20℃,沉淀的盐类通过抽气滤出,并用5 ml的水洗涤。在后续阶段(参见实施例5)中使用该产物的水溶液(102.1 g;最多172.7 mmol)。
出于分析目的,浓缩并干燥样品。
实施例5
2-氟-3-(吗啉-4-基)丙烯醛
方法A:
将实施例4的化合物的水溶液(最多251.5 mmol)加热至75℃。随后,逐滴加入43.8 g (503 mmol) 的吗啉和76.3 g (755 mmol)的三乙胺。在75℃下搅拌该混合物2 h并冷却至23℃,加入290 ml的二氯甲烷和100 ml的三乙胺。分离各相,用290 ml 二氯甲烷和100 ml三乙胺的混合物洗涤水相,过滤合并的有机相,用250 ml的饱和碳酸钾水溶液洗涤并在旋转蒸发器上在40℃下浓缩。加入 50 ml的甲苯,并进一步浓缩混合物。由此得到 34.2 g (理论值的81.9%) 的标题化合物。
方法B:
将43.8 g (503 mmol)吗啉和76.3 g (755 mmol)三乙胺的混合物加热至75℃,并在25分钟内逐滴加入实施例4 的化合物的水溶液(最多251.5 mmol)。随后,在75℃下搅拌混合物2 h并冷却至23℃,加入290 ml的二氯甲烷和100 ml的三乙胺。过滤混合物,分离各相,用290 ml 二氯甲烷和100 ml三乙胺的混合物洗涤水相,合并的有机相用250 ml的饱和碳酸钾水溶液洗涤并在旋转蒸发器上在40℃下浓缩。加入 50 ml的甲苯,并进一步浓缩。由此得到35.3 g (理论值的83.4%) 的标题化合物。
方法C:
将30.2 g (345.3 mmol)吗啉和52.5 g (518.0 mmol)三乙胺的混合物加热至75℃,并在75-80℃下逐滴加入实施例4方法B 的化合物的水溶液(最多172.7mmol)。将混合物在回流下搅拌2 h,冷却至23℃,并用100 ml的二氯甲烷洗涤。水相用100 ml二氯甲烷和15 ml三乙胺的混合物洗涤二次,合并的有机相用85 ml的饱和碳酸钾水溶液洗涤并在真空下在45-50℃下浓缩。加入 120 ml的甲苯,并将60 ml的甲苯蒸馏出。悬浮液在室温下搅拌过夜,产物通过抽气滤出,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此得到19.2 g (理论值的68.3%) 的标题化合物。
实施例6
5-氟-1-(2-氟苄基)-1H-吡唑并[3,4-b]吡啶-3-甲酸乙酯
方法A:
先将22.3 g (84.8 mmol)的5-氨基-1-(2-氟苄基)-1H-吡唑-3-甲酸乙酯(通过WO 00/06569实施例20A中所描述的方法制备)加入59.5 ml的乙醇中,并在RT下加入11.0 ml (169.6 mmol) 的甲磺酸、9.0 g (212.1 mmol) 的氯化锂和15.0 g (84.8 mmol)的实施例5的化合物。将混合物在回流温度下搅拌4.5 h。冷却至室温后,产物通过抽气滤出,用 4.5 ml的乙醇洗涤二次并与325 ml的水搅拌1 h。固体通过抽气滤出,用11.5 ml的水洗涤二次,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此得到 21.8 g (理论值的81.0%)的标题化合物。
方法B:
先将27.0 g (635.2 mmol)的氯化锂和42.2 g (254.1 mmol) 的实施例5 的化合物加入75 ml的乙醇中并加热至回流温度。在此温度下,在10分钟内加入66.9 g (254.1 mmol)的5-氨基-1-(2-氟苄基)-1H-吡唑-3-甲酸乙酯(通过WO 00/06569实施例20A中所描述的方法制备)和33.0 ml (508.2 mmol) 甲磺酸在180 ml乙醇中的溶液。将混合物在回流温度下搅拌2 h,然后加入120 ml 的异丙醇,将混合物冷却至62℃,将0.6 g的标题化合物用作晶种并将混合物在4 h内冷却至5℃ 。产物通过抽气滤出,与120 ml的异丙醇搅拌,抽气过滤,用180 ml的水洗涤,与300 ml的水搅拌0.5 h,抽气过滤,用300 ml的水洗涤,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得65.1 g (理论值的80.7%) 的标题化合物。
方法C:
先将5.42 g (20.6 mmol) 的5-氨基-1-(2-氟苄基)-1H-吡唑-3-甲酸乙酯(通过WO 00/06569实施例20A中所描述的方法制备)加入20 ml的乙醇中,并导入1.5 g (41.1 mmol) 的氯化氢。在10分钟内在回流温度下,将该溶液中计量加入到3.42 g (20.6 mmol) 的实施例5化合物在50 ml的乙醇中的溶液。将该混合物在回流温度下搅拌2 h,然后加入10 ml的异丙醇,并将该混合物冷却至5℃。产物通过抽气滤出,用10 ml的异丙醇洗涤,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得4.84 g (理论值的74.2%) 的标题化合物。
实施例7
5-氟-1-(2-氟苄基)-1H-吡唑并[3,4-b]吡啶-3-甲酰胺
将10 ml的乙醇、14.9 ml (441.2 mmol) 的甲酰胺和3.6 g (66.2 mmol) 的甲醇钠在甲醇中的溶液 (30%) 加入到在实施例6中获得的7.0 g (22.1 mmol)化合物中. 将反应混合物加热至95-100℃并将低沸物蒸馏出。混合物在125℃下搅拌1.5 h,加入30 ml 的水,冷却至室温以及搅拌1 h。沉淀的固体通过抽气滤出,每次用 8.5 ml的水洗涤三次,并在真空干燥箱中在 45℃以及温和的氮气流下干燥。由此获得6.2 g (理论值的97.5%) 的标题化合物。
实施例8
5-氟-1-(2-氟苄基)-1H-吡唑并[3,4-b]吡啶-3-甲腈
将17.3 g (60.0 mmol) 在实施例7中获得的化合物在40.5 ml环丁砜和5.4 ml乙腈中加热至103-107℃。之后,缓慢逐滴加入6.9 g (45.0 mmol)的氧氯化磷,同时搅拌,用2.8 ml的乙腈冲洗滴液漏斗,然后在107℃下搅拌该混合物1.5 h,直至转化完成(HPLC)。之后,将混合物冷却至室温并逐滴加入2.8 ml的环丁砜/乙腈(5:1 v/v),然后是17.8 ml的水。搅拌混合物0.5 h,逐滴加入 9.4 g 氨水(28%)在22.7 ml水中的溶液并另外再搅拌2 h。沉淀的固体通过抽气滤出,每次用20.5 ml的水洗涤三次,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得14.7 g (理论值的91.9%) 的标题化合物。
实施例9
5-氟-1-(2-氟苄基)-1H-吡唑并[3,4-b]吡啶-3-甲酰亚胺酰胺-盐酸盐
将406.0 g (1.50 mol)实施例8的化合物悬浮在2.08 l 的乙醇中。随后,加入54.1 g (0.30 mol) 甲醇钠在甲醇中的溶液(30%),并在室温下搅拌该混合物过夜。 加入88.4 g (1.65 mol)的氯化铵,将混合物加热至65℃并在65℃下搅拌3.5 h。蒸馏出溶剂并将残余物和 1.6 l的乙酸乙酯搅拌过夜。沉淀的固体通过抽气滤出,每次用140 ml的乙酸乙酯洗涤二次,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得 441.4 g (理论值的90.7%) 的标题化合物。
实施例10
[(E)-苯基二氮烯基]丙二腈
方法A:
在0-5℃下将262 g的浓盐酸(2.59 mol)和117.5 ml的水逐滴加至1525 ml的水和117.5 g (1.26 mol)的苯胺中。随后,在1 h内逐滴加入87.1 g (1.26 mol)亚硝酸钠在222.5 ml的水中的溶液,并用60 ml的水冲洗,和在0-5℃下搅拌该混合物15分钟。之后,在此温度下,在45分钟内逐滴加入131.4 g (1.60 mol)乙酸钠在665 ml水(19 ml)中的溶液,并用 60 ml的水冲洗,在1 h内逐滴加入83.4 g (1.26 mol) 丙二腈在233 ml乙醇中的溶液。对其使用68.5 ml的乙醇冲洗,并在0-5℃下搅拌该混合物2 h。黄色固体通过抽气滤出,并每次用625 ml的水和488 ml的冷甲苯洗涤三次。将仍潮湿的残余物溶解在872 g的DMF中。由此获得1117.0 g标题化合物的DMF溶液。
方法B:
在0-5℃下将87.4 g的浓盐酸 (0.86 mol)和39.5 ml的水逐滴加至508.5 ml的水和39.2 g (0.42 mol)的苯胺中。随后,在1 h内逐滴加入29.0 g (0.42 mol)亚硝酸钠在74.5 ml的水中的溶液,并用20 ml的水冲洗,和在0-5℃下搅拌该混合物15分钟。之后,在此温度下,在45分钟内逐滴加入43.8 g (0.54 mol)乙酸钠在221.5 ml水中的溶液,并用 20 ml的水冲洗,在1 h内逐滴加入27.8 g (0.42 mol)丙二腈在77.5 ml乙醇中的溶液。对其使用23 ml的乙醇冲洗,并在0-5℃下搅拌该混合物2 h。黄色固体通过抽气滤出,并每次用208.5 ml的水和162.5 ml的冷甲苯洗涤三次。获得103.1 g潮湿的产物。将13.8 g潮湿的产物溶解在13.9 g的环丁砜中。由此获得27.7 g标题化合物的环丁砜溶液。
实施例11
2-[5-氟-1-(2-氟苄基)-1H-吡唑并[3,4-b]吡啶-3-基]-5-[(E)-苯基二氮烯基]嘧啶-4,6-二胺
方法A:
将448.2 g (1.38 mol) 实施例9的化合物悬浮在1059 ml的DMF中。将混合物加热至85℃,并在此温度下逐滴加入212 ml (1.52 mol) 的三乙胺。随后,在20分钟内逐滴加入1751 g实施例10的DMF溶液,并用490 ml的DMF冲洗,和在100℃下搅拌该混合物过夜。将反应混合物冷却至RT,逐滴加入656 ml的水,并将混合物在RT下搅拌0.5 h,然后冷却至0-5℃并另外再搅拌1 h。固体通过抽气滤出,洗涤两次,每次用1443 g水和236 g甲醇的溶液,然后用586 ml 的甲醇洗涤,抽气干燥并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得522.2 g (理论值的82.5%) 的标题化合物。
方法B:
将30.0 g (92.7 mmol)实施例9的化合物悬浮在72 ml的DMF中。将混合物加热至100℃,并在此温度下在30分钟内逐滴加入14.2 ml (101.9 mmol)的三乙胺和150 g实施例10的DMF溶液的混合物。将其使用30 ml的DMF冲洗,并在100℃下搅拌该混合物20 h。将反应混合物冷却至95-90℃,在10分钟内逐滴加入24 ml的水,然后在1.5 h内将混合物冷却至0-5℃并再搅拌1 h。固体通过抽气滤出,用60 g的水和60 g的二甲基甲酰胺的溶液洗涤,每次用50 g水和50 g甲醇的溶液洗涤二次,然后用40 ml的甲醇洗涤,抽气干燥,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得35.5 g (理论值的83.7%) 的标题化合物。
方法C:
将11.7 g (36.0 mmol)实施例9的化合物悬浮在15.6 ml的环丁砜中。将混合物加热至100℃,并在此温度下在35分钟内逐滴加入5.5 ml (39.6 mmol)的三乙胺和27.7 g实施例10方法B的环丁砜溶液的混合物。将其使用2 ml的环丁砜冲洗,并在100℃下搅拌该混合物2.5 h。将反应混合物冷却至60℃,逐滴加入90 ml的异丙醇,然后在15分钟内将混合物冷却至0-5℃并再搅拌2.5 h。固体通过抽气滤出,洗涤三次,每次用50 g的水和24 ml的异丙醇,抽气干燥,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得14.2 g (理论值的85.9%) 的标题化合物。
实施例12
2-[5-氟-1-(2-氟苄基)-1H-吡唑并[3,4-b]吡啶-3-基]嘧啶-4,5,6-三胺
方法A:
先将182.0 g (0.39 mol)实施例11的化合物加入1.82 l的DMF中,然后加入 4.2 g 的钯 (5%在炭上,50% 水-湿度)。 在60℃和60 巴氢气压下进行氢化,同时搅拌过夜。将混合物通过硅藻土过滤,并用150 ml的DMF洗涤,和然后用150 ml的甲醇洗涤,并在60-70℃下浓缩至425 g重量的蒸馏残余物。将残余物加热至75-80℃,在此温度下逐滴加入300 ml的甲醇,并搅拌该混合物15分钟。在1 h内将该混合物冷却至RT,然后逐滴加入1290 ml的水,并搅拌混合物过夜。固体通过抽气滤出,每次用500 ml水洗涤二次,抽气干燥,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得159.7 g 的标题化合物。产物具有73.7重量%的含量和12.4重量%的DMF(理论值的80.3%),并由此直接用在后续阶段中。根据水洗的强度,DMF含量在10-17重量%的范围内。
方法B:
将25.0 g来自方法A的含DMF的固体悬浮在220 ml的水中,并通过抽滤器用抽气过滤。固体在抽滤器上洗涤四次,每次用100 ml的95℃的水,抽气干燥,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得21.2 g的无DMF的标题化合物。
出于分析的目的,通过硅胶过滤的方法纯化样品:
。
实施例13
{4,6-二氨基-2-[5-氟-1-(2-氟苄基)-1H-吡唑并[3,4-b]吡啶-3-基]嘧啶-5-基}氨基甲酸甲酯
方法A:
将4.0 g (77.0重量%,8.36 mmol)实施例12的化合物在37.9 ml 异丙醇中加热至35℃,然后逐滴加入0.84 ml (10.87 mmol) 的氯甲酸甲酯。在35-40℃下搅拌该混合物20 h,并加热至50℃,以及加入9.5 ml的甲醇。随后,在0.5 h内,逐滴加入1.9 ml的三乙胺并用1.3 ml的甲醇冲洗,和在50℃下搅拌该混合物1 h。之后,将反应混合物冷却至RT,并在RT下搅拌1 h,固体通过抽气滤出,每次用8 ml乙醇洗涤三次,抽气干燥并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得3.4 g的粗产物。将3.0 g的粗产物置于8 ml的DMSO中搅拌5分钟,加入13.0 ml的乙酸乙酯和50 mg的活性炭,并将混合物加热回流(84℃)15分钟。将此悬浮液热过滤并用1.9 ml的乙酸乙酯洗涤过滤残余物1)。将60 ml的乙酸乙酯和16 ml的乙醇加热至60℃,并逐滴加入合并的滤液,在60℃下再搅拌1.5 h。在25分钟内将悬浮液冷却至RT,再搅拌1.5 h,之后冷却至0-5℃,并另外再搅拌1 h。固体通过抽气滤出,每次用6.4 ml的乙酸乙酯洗涤二次,抽气干燥并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此得到2.2 g (理论值的70.0%) 的标题化合物。
1)根据所述的制备方法,在此时得到二-二甲基亚砜-溶剂合物,其通过在X射线衍射图中的反射和IR光谱中的谱带表征在表2和表4中。
式(I)化合物的二-二甲基亚砜-溶剂合物比现有技术中的物质具有明显更好的可过滤性的优点。此外,通过式(I)化合物的二-二甲基亚砜-溶剂合物的制备方法获得了非常高纯度的式(I)的化合物。
方法B:
将4.0 g (10.8 mmol) 实施例12 方法B的化合物在37.9 ml的异丙醇中加热至35℃,然后逐滴加入1.1 ml (14.1 mmol)的氯甲酸甲酯。在35-40℃ 下搅拌16.5 h并冷却至RT,加入2.1 ml的氨水 (28%)。随后加入4.2 ml的水,并搅拌该混合物2.5 h。固体通过抽气滤出,每次用5 ml的水洗涤二次,抽气干燥并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得4.4 g的粗产物。
方法C:
将4.0 g (10.8 mmol) 实施例12 方法B的化合物在37.9 ml的异丙醇中加热至35℃,然后逐滴加入1.1 ml (14.1 mmol)的氯甲酸甲酯。在35-40℃ 下搅拌该混合物16.5 h,并在50 ℃下加入9.5 ml的甲醇。随后,在20分钟内逐滴加入2.42 ml的三乙胺并用1.3 ml的甲醇冲洗,在50 ℃下搅拌该混合物1 h。之后,将反应混合物冷却至RT,并在RT下搅拌1 h,固体通过抽气滤出,每次用8 ml的甲醇洗涤三次,抽气干燥并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得4.3 g的粗产物。
方法D:
将6.9 g的粗产物在18.4 ml的DMSO中搅拌5分钟,加入30.0 ml的乙酸乙酯和115 mg活性炭并加热回流该混合物(84℃)15分钟。热过滤悬浮液并用4.4 ml的乙酸乙酯洗涤过滤器残余物。将138 ml的乙酸乙酯加热至50 ℃,逐滴加入合并的滤液并在45-50℃下再搅拌1 h。将悬浮液在1.5 h内冷却至0-5℃,并另外再搅拌1 h。固体通过抽气滤出,每次用14.8 ml的乙酸乙酯洗涤二次和抽气干燥1 h。得到6.4 g的二-二甲基亚砜-溶剂合物潮湿产物1)。
方法E:
将2.0 g的二-二甲基亚砜-溶剂合物在回流温度下在 40 ml乙酸乙酯和11.1 ml 乙醇中搅拌17 h,冷却至RT,并另外再搅拌1 h。固体通过抽气滤出,每次用1.4 ml乙酸乙酯洗涤四次,并在真空干燥箱中在50℃以及温和的氮气流下干燥。由此获得1.4 g 以多晶型物I存在的标题化合物。
方法F:
将0.5 g的二-二甲基亚砜-溶剂合物在回流温度下在12.5 ml的溶剂中搅拌17 h,冷却至RT并另外再搅拌1 h。固体通过抽气滤出,用2 ml溶剂洗涤并抽气干燥30分钟。由此获得0.3 g以多晶型物I存在的标题化合物。
使用以下溶剂:
1.) 9 ml 乙酸乙酯/3.5 ml乙醇/0.3 ml水
2.) 12.5 ml 异丙醇
3.) 12.5 ml 异丙醇/0.3 ml水
4.) 12.5 ml 甲醇
5.) 12.5 ml 甲醇/0.3 ml水
6.) 12.5 ml 乙腈
7.) 12.5 ml 丙酮
8.) 12.5 ml 四氢呋喃
9.) 12.5 ml 甲基叔丁基醚。
表1显示了X射线衍射图的反射。表3显示了IR光谱的谱带。
以结晶多晶型物I存在的化合物(I)以较高的稳定性而引人注目,更特别地是,其在微粒化过程中仍是稳定的,因此不会发生转化和重结晶。
式(I)的化合物可通过上述方法来制备。由此提供的以结晶多晶型物存在的式(I)的化合物在下文中被称为多晶型物I。多晶型物I具有257℃的熔点,在特性X射线衍射图中具有(2 θ) 5.9、6.9、16.2、16.5、24.1和24.7的特征反射,并且在特性IR光谱中具有(用cm-1表示) 1707、1633、1566、1475、1255和1223的特征最大谱带 (表1和3,图1和5)。
令人惊讶的是,发现式(I)化合物的四种其它的多晶型物、单水合物、二水合物、DMF/水-溶剂合物和二-二甲基亚砜-溶剂合物,以及三乙酸-溶剂合物。以多晶型物II存在的式(I)的化合物在约253℃熔化;以多晶型物III存在的式(I)的化合物具有约127℃的熔点。式(I)的化合物的多晶型物IV在246℃的温度熔化,而多晶型物V具有234℃的熔点。单水合物含有约4.1% 水,二水合物含有7.8% 水,DMF/水-溶剂合物含有13.6% 二甲基甲酰胺和0.9 % 水,二-DMSO-溶剂合物含有26.8%二甲基亚砜,而三乙酸-溶剂合物含有29.7%乙酸酯。所提及的每种结晶形式具有各自的特征X射线衍射图和IR光谱(表2和3,图1 – 4,6 - 14)。
表1:多晶型物I至V的X射线衍射
表2:多晶型物水合物和溶剂合物的X射线衍射
表3:多晶型物I至V的IR光谱
表4:水合物和溶剂合物的IR光谱
图1: 以多晶型物I、II和III存在的式(I)的化合物的IR光谱
图2: 以多晶型物IV、V存在和作为三乙酸-溶剂合物的式(I)的化合物的IR光谱
图3: 作为二-DMSO-溶剂合物、DMF/水-溶剂合物和单水合物的式(I)的化合物的IR光谱
图4: 作为二水合物的式(I)的化合物的IR光谱
图5: 以多晶型物I存在的式(I)的化合物的X射线衍射图
图6: 以多晶型物II存在的式(I)的化合物的X射线衍射图
图7: 以多晶型物III存在的式(I)的化合物的X射线衍射图
图8: 以多晶型物IV存在的式(I)的化合物的X射线衍射图
图9: 以多晶型物V存在的式(I)的化合物的X射线衍射图
图10: 作为三乙酸-溶剂合物的式(I)的化合物的X射线衍射图
图11: 作为二-DMSO-溶剂合物的式(I)的化合物的X射线衍射图
图12: 作为DMF-水-溶剂合物的式(I)的化合物的X射线衍射图
图13: 作为单水合物的式(I)的化合物的X射线衍射图
图14: 作为二水合物的式(I)的化合物的X射线衍射图。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种N‑取代吡唑并[3,4‑d]嘧啶酮类化合物及制备方法与应用与流程
- 取代吡唑酰胺类化合物及其应用的制造方法与工艺
- 取代的吡唑并[1,5‑a]嘧啶以及它们在治疗医学障碍中的用途的制造方法与工艺
- 取代的吡唑并[1,5‑a]嘧啶以及它们在治疗医学障碍中的用途的制造方法与工艺
- 取代二氢吡咯并吡唑化合物的制造方法与工艺
- 取代的吡唑并[1,5‑a]吡啶和咪唑并[1,2‑a]吡嗪及其用途的制造方法与工艺
- 经取代的吡唑基吡唑衍生物及其作为除草剂的用途的制造方法与工艺
- 经取代的吡唑基吡唑衍生物及其作为除草剂的应用的制造方法与工艺
- 经取代的吡唑基吡唑衍生物及其作为除草剂的用途的制造方法与工艺
- 经取代的吡唑基吡唑衍生物及其作为除草剂的应用的制造方法与工艺
- 一种4‑氨基安替比林产品的合成工艺及其试液的配备方法与制造工艺
- 一种吡唑酮生产工艺的制造方法与工艺
- 一类含呋喃的苯基联吡唑甲酰胺类衍生物及其制备方法和用图
- N’?5?(取代苯基)?2?呋喃甲酰基?5?丙基?1h?吡唑?3?甲酸乙酯类化合物及其制备方法和应用
- 吡唑啉酮化合物及其用图
- 5-(4-氟苯基)-N–吗啉基-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 5-(4-氟苯基)-N–(4-氯苯基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 含醚性氧原子的全氟烷基取代吡唑环化合物及其制造方法
- 5-(4-氟苯基)-N–(4-氯苯乙基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 含有色酮结构的吡唑类n‐对硝基苯基马来酰亚胺衍生物及其制备方法与应用