皮考啉腙酸衍生物双氧钒(V)配合物及其制备方法与流程
本发明涉及金属配合物,具体涉及一种皮考啉腙酸衍生物双氧钒(V)配合物及其制备方法和应用。
背景技术:
蛋白酪氨酸磷酸酶(PTPs)是生物体内细胞信号转导过程中发挥重要作用的一类酶,它能够特异性去除蛋白酪氨酸残基上的磷酸基团,从而广泛影响细胞生长、分化、迁移、凋亡和免疫应答等过程。蛋白酪氨酸磷酸酶1B(PTP1B)是第一个被发现和纯化的PTP酶,它通过对胰岛素受体及其底物的酪氨酸残基去磷酸化,从而对胰岛素信号转导过程进行负调控。研究表明敲除PTP1B基因的小鼠对胰岛素的敏感性和葡萄糖的耐受性都明显增强并且正常生长。目前PTP1B被公认为是研发治疗Ⅱ型糖尿病药物的作用靶点。因此,寻找与PTP1B进行特异性结合和作用的小分子,研究小分子和PTP1B相互作用,则成为研发以PTP1B为靶点的新药的基础。
钒配合物具有类胰岛素的作用,并且已经在糖尿病的治疗中表现出了一定的效果。研究表明钒配合物的类胰岛素作用与其对PTP1B的抑制有关。目前除BMOV外,一些氧钒配合物也具有类胰岛素性质,但是配位形式为VO2(NNO)的配合物类胰岛素性质报道较少,并且对于双氧钒配合物对细胞内PTP1B活性的抑制的研究很少,所以我们制备并研究了基于PTP1B靶点的具有类胰岛素性质的皮考啉腙酸衍生物双氧钒(V)配合物,为糖尿病的治疗提供了一种新的药物开发途径。
技术实现要素:
本发明的目的在于提供一种皮考啉腙酸衍生物双氧钒(V)配合物及其制备方法,以及作为一
种PTP1B选择性抑制剂和在制备抗糖尿病药物中的应用。
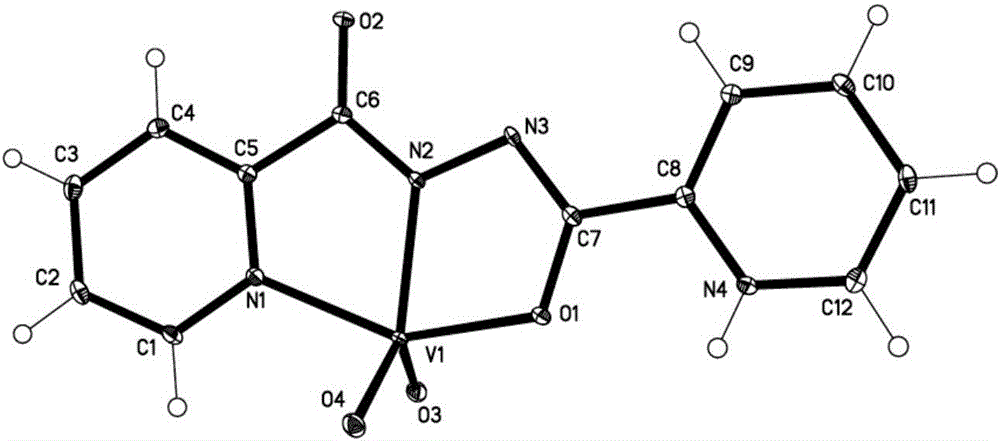
本发明提供的一种皮考啉腙酸衍生物双氧钒(V)配合物,分子式为:VO2(HPPCH),其中H2PPCH为N'-皮考啉基皮考啉腙酸(N'-picolinoylpicolino-hydrazonic acid),该配合物结构式为:
本发明提供的一种皮考啉腙酸衍生物双氧钒(V)配合物的制备方法,包括如下步骤:
(1)将VOSO4溶解于蒸馏水中,将上述溶液滴加到N'-(吡啶-2-亚甲基)吡啶甲酰肼的甲醇溶液中,VOSO4与N'-(吡啶-2-亚甲基)吡啶甲酰肼的摩尔比为1-2:1,优选2:1;
(2)室温搅拌8-10小时,过滤,得到红棕色滤液,室温缓慢挥发,3天后有红棕色块状晶体析出。
本发明制备的皮考啉腙酸衍生物双氧钒(V)配合物的晶体属于单斜晶系,空间群为P21/c,晶胞参数为:α=90.00°,β=109.44(3)°,γ=90.00°。
X射线粉末衍射证实该配合物晶体样品均一稳定。电喷雾质谱表明该配合物在溶液中能够稳定存在,其组分与固体一致。半数抑制浓度(IC50)测定证明该配合物能够高效且选择性地抑制蛋白酪氨酸磷酸酶1B的活性,蛋白免疫印迹实验表明该配合物能够强烈地增强人肝癌细胞(HepG2)内PTP1B底物的磷酸化水平,从而证明该配合物能够有效抑制人肝癌细胞(HepG2)内PTP1B的活性。细胞毒性实验表明该配合物的细胞毒性低于VOSO4。因此,本发明的配合物不仅可用作高效低毒的蛋白酪氨酸磷酸酶1B的抑制剂,而且可以在制备抗糖尿病药物中应用。
本发明的优点和效果:
本发明的配合物生产成本较低,合成方法简单,过程易控制,配合物稳定,晶体易得,产率高,能够强烈抑制人肝癌细胞(HepG2)内的蛋白酪氨酸磷酸酶1B的活性,具有比VOSO4更优越的抑制细胞内蛋白酪氨酸磷酸酶1B的活性的性能,且细胞毒性小于VOSO4,为糖尿病治疗提供了一种新的药物开发途径。
附图说明
图1本发明配合物VO2(HPPCH)的晶体结构图。
图2本发明配合物VO2(HPPCH)在298K的X射线粉末衍射图(实验及模拟图)。
图3本发明配合物VO2(HPPCH)的电喷雾质谱图。
图4本发明配合物VO2(HPPCH)抑制PTPs活性的IC50值测定曲线。
图5本发明配合物VO2(HPPCH)、VOSO4、配体对人肝癌细胞(HepG2)内PTP1B底物的影响。
图6本发明配合物VO2(HPPCH)对人肝癌细胞(HepG2)细胞活力的影响。
具体实施方式
下面结合附图和实施例,对本发明作进一步详细说明。
实施例1.本发明配合物VO2(HPPCH)的制备及晶体培养方法。
称取0.049g(0.3mmol)VOSO4溶解于5mL蒸馏水中,将上述溶液滴加到0.034g(0.15mmol)N'-(吡啶-2-亚甲基)吡啶甲酰肼的10mL甲醇溶液中,室温搅拌8小时,过滤,得到红棕色滤液,室温缓慢挥发,3天后有红棕色块状晶体析出,抽滤,分别用水、甲醇、乙醚洗涤三次,真空干燥,产率为60%。元素分析:理论值:C44.46,H 2.80,N17.28;实验值:C 44.37,H 2.94,N 17.16。
实施例2.本发明配合物VO2(HPPCH)的晶体结构测定和结果。
晶体结构测定采用北京同步辐射源,使用MARCCD-165探测器收集衍射数据数据经HKL2000还原后,用SHELXL-97直接发解得晶体结构,C原子采用理论加氢,N原子通过差值傅里叶图加氢,详细的晶体测定数据见表1。结构见图1。
表1配合物的晶体学数据
实施例3.本发明配合物VO2(HPPCH)的粉末衍射。
X-射线粉末衍射结果表明,晶体样品物相均一,实验衍射图谱与依据晶体结构模拟的粉末衍射图谱一致,见图2。
实施例3.本发明配合物VO2(HPPCH)的电喷雾质谱。
为了研究配合物在溶液中的存在形式,将适量的配合物固体粉末溶于甲醇中,振荡使其溶解达到饱和,并离心得到澄清透明溶液,上样至电喷雾质谱仪,采用电喷雾离子源,以正离子方式检测样品并记录质谱数据。图3是配合物的电喷雾质谱图,在电喷雾质谱图中能观察到配合物相应的分子离子峰。表2是配合物的正离子电喷雾质谱归属。结果表明,该结果与元素分析所得结果一致,表明配合物在溶液中以我们设计的结构形式稳定存在。
表2配合物的正离子电喷雾质谱归属
实施例4.本发明配合物VO2(HPPCH)对PTP1B活性抑制及其选择性测定。
IC50测定原理:
IC50即半数抑制浓度,是引起酶活性下降至原酶活性一半时所需抑制剂的浓度,IC50值是用来评判抑制剂抑制能力大小的标准。其数值越低,表明抑制剂的抑制能力越强。蛋白酪氨酸磷酸酶(PTPs)能使反应底物对硝基苯磷酸二钠盐(pNPP)分解成黄色的对硝基苯酚(pNP),该产物在405nm处有很强的吸收。PTPs与配合物作用后,通过检测405nm处吸光度值的变化来间接检测酶活性的变化情况。
IC50的测定方法:
进行IC50值测定实验时,首先将抑制剂配制成10-2M的DMSO母液,然后稀释到一系列梯度的适宜浓度,用现配的缓冲溶液溶解PTPs使其浓度适宜,备用。抑制剂抑制PTPs的活性实验是在96孔板中进行,前三排加入83μL含酶的MOPS缓冲溶液(50mM NaCl,20mM MOPS,pH=7.2),最后一排加入不含酶的MOPS缓冲溶液作为对照。再以列为单位,依次加入10μL不同浓度梯度的抑制剂,置于37℃恒温水浴锅反应30分钟后,向上述溶液加入2μL pNPP(0.1M)来启动反应,15分钟后,用5μL(2M)NaOH终止反应,通过酶标仪测定底物的分解产物pNP在405nm处各孔的紫外吸收值。以配合物浓度的对数作为横坐标,抑制率作为纵坐标,用Origin程序作图,拟合得到配合物对酶抑制能力的曲线,从而求得IC50值。实验重复三次以上。
实验结果:本发明配合物对PTP1B,TCPTP,SHP-1,SHP-2的半抑制浓度(IC50)分别为0.13μM,0.98μM,2.02μM和16.26μM。配合物对PTP1B的抑制能力分别是TCPTP,SHP-1,SHP-2的7,15和125倍。由此可见,该配合物能够有效地抑制PTP1B活性,并且对PTP1B有较好的选择性,见图4。
实施例5.本发明配合物VO2(HPPCH)对人肝癌细胞(HepG2)内PTP1B活性抑制的测定。
蛋白免疫印迹法测定原理:
蛋白免疫印迹法的基本原理是通过特异性抗体对凝胶电泳处理过的细胞或生物组织样品进行着色。通过分析着色的位置和着色深度获得特定蛋白质在所分析的细胞或组织中表达情况的信息。配合物作用细胞后,通过检测细胞内PTP1B底物的表达量变化来间接检测细胞内PTP1B活性的变化情况。
蛋白免疫印迹法步骤:
(1)样品处理。将人肝癌细胞(HepG2)在含有10%胎牛血清的培养基中于37℃、5%CO2培养箱中培养,待细胞处于对数增长期,向培养皿内加药。实验组分别加入终浓度为0.1、1、5、10μM的配合物VO2(HPPCH),10μM VOSO4、10μM配体(N'-(吡啶-2-亚甲基)吡啶甲酰肼),混合溶剂(DMSO(10%)+0.85%NaCl(90%)),对照组为空白细胞,继续培养48h,7000rpm离心收集细胞,用裂解液在冰上裂解细胞50分钟后,13000rpm离心10-15分钟,缓慢吸取上清,BCA法测定蛋白浓度,分装蛋白质(每管80μg),煮沸使蛋白质变性,准备上样。
(2)电泳分离。首先制备电泳凝胶(10%分离胶和5%浓缩胶),将(1)中处理好的样品及蛋白marker分别上样于相应泳道,进行电泳分离。
(3)蛋白的膜转移。a电泳结束后将胶条切至合适大小,用电转缓冲液平衡10分钟;b预先裁好与胶条同样大小的滤纸和PVDF膜,浸入电转缓冲液中平衡10分钟;c转膜,转膜装置从下至上一次是阳极板-海绵-5层滤纸-PVDF膜-胶条-5层滤纸-海绵-阴极板。接通电源,恒压110V,电转2小时。
(4)免疫杂交与显色-蛋白检测。a封闭。电转结束后,将PVDF膜用封闭液(0.5g/10mL脱脂奶粉)室温封闭30分钟;b将PVDF膜平整地放入杂交袋中,取2μL一抗加入1.0mL封闭液,混匀,加入杂交袋中PVDF膜印有蛋白质的一侧,平放,4℃过夜。本实验中所用的一抗分别为p-Src(Y529),p-EGFR(Y1029),p-IRS-1(Y896)和(IR/IGF1R)[pYpYpY1158/1162/1163],GADPH为内参;c TBST洗脱液洗脱PVDF膜3次,取二抗3μL加入1.5mL封闭液中,接二抗,室温孵育1小时,用TBST洗脱液洗脱PVDF膜3次,准备曝光。本实验中用的二抗为Goat anti-Rabbit IgG(H&L)-HRP;d将PVDF膜放置在暗盒内,滴加发光液,反应1-2分钟,在暗室内将X光片覆盖在膜上,盖好暗盒,曝光约5分钟,显影,定影。
实验结果:随着配合物浓度的增大,细胞内PTP1B底物(p-Src(Y529),p-EGFR(Y1029),p-IRS-1(Y896)和(IR/IGF1R)[pYpYpY1158/1162/1163])的磷酸化水平逐渐增强,并且本发明配合物的性能优于阳性对照组VOSO4,表明本发明配合物能够强烈抑制细胞内PTP1B的活性,见图5。
实施例6.本发明配合物VO2(HPPCH)对人肝癌细胞(HepG2)细胞毒性的测定。
MTT法测定原理:MTT全称3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide,商品名为噻唑蓝,是一种黄颜色的染料,用于检测细胞存活和增殖。外援性的MTT可透过细胞膜进入细胞,与活细胞线粒体中的琥珀酸脱氢酶发生氧化还原反应生成不溶于水的蓝紫色结晶甲瓒,并沉积在细胞培养皿底部。而死细胞中不含琥珀酸脱氢酶,因此加入MTT不会生成甲瓒。生成的甲瓒结晶能溶于二甲基亚砜,显紫色,用酶标仪检测490nm波长处溶液的吸收度值可间接反应活细胞数量。在一定细胞数范围内,甲瓒结晶形成的量与活细胞数成正比。
MTT法步骤:将人肝癌细胞(HepG2)在含有10%胎牛血清的培养基中于37℃、5%CO2培养箱中培养,用MTT法检测了本发明皮考啉腙酸衍生物双氧钒(V)配合物对肝癌细胞(HepG2)的毒性,同时以VOSO4为阳性对照,检测本发明配合物随时间和浓度变化对细胞活力的影响。实验取对数生长期的HepG2细胞经消化、离心收集后配制成1mL的细胞悬液,由血球计数板测得细胞悬液内的细胞个数,取适量体积的该细胞悬液将其稀释成细胞密度约为1×10-4个/mL的细胞悬液,均匀接种于96孔板,每孔200μL,将96孔板放置在CO2培养箱中孵化,至细胞单层铺满孔低,加入4个浓度梯度的配合物,每组设6个复孔。细胞继续在CO2培养箱中分别培养孵化72小时后,倒置显微镜下观察细胞形态变化,然后每孔加入20μL MTT(5mg/mL)溶液,继续孵育4小时,小心吸去上清液,每孔加入200的DMSO溶液,振荡10min,待底部蓝色结晶状固体完全溶解,用酶标仪测得各孔溶液490nm处的吸光度值A。
以药物浓度为横坐标,细胞存活率为纵坐标绘制细胞生长曲线。
细胞存活率(%)=(A实验组/A对照组)×100
实验结果:图6为相同浓度梯度的皮考啉腙酸衍生物双氧钒(V)配合物和VOSO4分别与HepG2细胞作用72小时后,它们对细胞活力的比较。实验结果表明,本发明配合物对人肝癌细胞(HepG2)有一定的毒性,但其毒性明显要小于VOSO4,因此,本发明配合物可用于PTP1B抑制剂和糖尿病治疗的潜在药物。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种5-[3-(2,5-二乙氧基-4-甲磺酰基-苄基)-脲基]-2-乙氧基-n-对甲苯基-苯甲酰胺新 ...的制作方法
- 一种5-[3-(2,5-二乙氧基-4-甲磺酰基-苄基)-脲基]-2-乙氧基-n-异丙基-苯甲酰胺新化 ...的制作方法
- 一种5-[3-(2,5-二乙氧基-4-甲磺酰基-苄基)-脲基]-2-乙氧基-n-丙基-苯甲酰胺新化合 ...的制作方法
- 一种5-[3-(2,5-二乙氧基-4-甲磺酰基-苄基)-脲基]-2-乙氧基-n-乙基-苯甲酰胺新化合 ...的制作方法
- 一种n-(4-乙氧基羰基苯基)-n’-甲基-n’-苯基甲脒中间体的生产系统的制作方法
- 一种n-[5-(4-溴苯基)-6-[2-[(5-溴-2-嘧啶基)氧]乙氧基]-4-嘧啶基]-n’-丙基磺酰胺 ...的制作方法
- [2-(1h)-喹啉酮-3-基]萘氨甲基膦酸酯作为抗癌药物的应用
- 富贵绒的染整工艺的制作方法
- 羊羔绒面料的制作方法
- α-(8-喹啉氧基)单取代酞菁锌治疗银屑病的用图