一种盐酸替匹嘧啶及其中间体6‑氯甲基尿嘧啶的合成方法与流程
本发明涉及医药技术领域,尤其涉及一种盐酸替匹嘧啶及其中间体6-氯甲基尿嘧啶的合成方法。
背景技术:
曲氟尿苷盐酸替吡嘧啶片(TAS-102),是由大鹏药业(Taiho Pharma)开发,于2014年日本MHLW批准,用于治疗不可切除型或复发型晚期结直肠癌的抗肿瘤复方药物。其中,盐酸替匹嘧啶(或盐酸替吡嘧啶)为抗肿瘤药物曲氟尿苷嘧啶片的主要成分。盐酸替匹嘧啶是胸苷磷酸化酶抑制剂,可有效减少在药物代谢过程中曲氟尿苷的降解。
目前,盐酸替匹嘧啶的主要合成策略包括:
1、以5-氯-6-甲基嘧啶为起始原料,首先用NCS(N-氯代丁二酰亚胺)进行甲基的氯代,再与2-氨基吡咯烷盐酸盐缩合得到产品;反应式如式1所示:
2、以6-氯甲基尿嘧啶为起始原料,首先经过磺酰氯氯代,再与2-氨基吡咯烷盐酸盐缩合,最后成盐得到产品;经过氯代、缩合的反应式如式2所示:
第一种合成策略的氯代收率为10%,缩合收率为38%,整体收率较低,生产过程操作困难,成本较高。相比第一种合成策略,第二种合成策略的收率更高,所得产品纯度更高,为目前主要用于盐酸替匹嘧啶合成的策略。
第二种合成策略所采用的起始物料为6-氯甲基尿嘧啶,现有技术一般采用6-甲基尿嘧啶为原料,依次经氧化、还原、氯代三个工艺步骤制得。目前,在该制备过程中氧化工艺所选用的氧化剂多为二氧化硒。但是,二氧化硒具有较高的毒性,增加了药品自身的安全隐患,不利于环境友好性。
因此,开发更适合医药工业使用的6-氯甲基尿嘧啶的合成方法具有重要的实践意义。
技术实现要素:
有鉴于此,本申请提供一种盐酸替匹嘧啶及其中间体6-氯甲基尿嘧啶的合成方法,本申请提供的6-氯甲基尿嘧啶的合成方法具有环境友好性,且产品收率及纯度高,利于制备盐酸替匹嘧啶等。
本发明提供一种6-氯甲基尿嘧啶的合成方法,包括以下步骤:
将6-甲基尿嘧啶与氧化铜在溶剂中进行氧化反应,得到6-甲酰基尿嘧啶;
还原6-甲酰基尿嘧啶,得到6-羟甲基尿嘧啶;
将6-羟甲基尿嘧啶经氯代反应,得到6-氯甲基尿嘧啶。
优选地,所述氧化铜与6-甲基尿嘧啶的摩尔比例为1:60~70。
优选地,所述氧化反应的温度为110~115℃。
优选地,所述氧化反应在溶剂乙酸中进行。
优选地,所述氧化反应的时间为5h~8h。
优选地,将氧化反应得到的混合物在温度为55~65℃且压力为0.09MPa的条件下,减压蒸馏,然后用硅藻土过滤,得到含6-甲酰基尿嘧啶的滤液。
优选地,将所述滤液调节pH值后再过滤,取滤饼醇洗,经60℃干燥7小时~8小时,得到固体6-甲酰基尿嘧啶。
优选地,采用硼氢化钠在水存在的条件下还原6-甲酰基尿嘧啶,得到6-羟甲基尿嘧啶。
优选地,在二甲基甲酰胺存在的条件下,6-羟甲基尿嘧啶与二氯亚砜进行氯代反应,得到6-氯甲基尿嘧啶。
本发明提供一种盐酸替匹嘧啶的合成方法,包括以下步骤:
将6-甲基尿嘧啶与氧化铜在溶剂中进行氧化反应,得到6-甲酰基尿嘧啶;
还原6-甲酰基尿嘧啶,得到6-羟甲基尿嘧啶;
将6-羟甲基尿嘧啶经氯代反应,得到6-氯甲基尿嘧啶;
将6-氯甲基尿嘧啶依次经过氯代、缩合、成盐,得到盐酸替匹嘧啶。
与现有技术相比,本发明采用金属氧化剂氧化铜代替了现有工艺使用的二氧化硒作为氧化剂,实现了6-甲基尿嘧啶氧化,而后经还原、氯代工艺制备了6-氯甲基尿嘧啶。按照本发明所述制备方法,所得6-氯甲基尿嘧啶的纯度>99.0%。本发明优化了盐酸替匹嘧啶中间体(6-氯甲基尿嘧啶)的合成方法,以低毒性的金属氧化剂代替了二氧化硒实现6-甲基尿嘧啶的氧化工艺,在增加环境友好性的同时,提高了6-氯甲基尿嘧啶的收率及纯度。
本发明采用上述工艺所得6-氯甲基尿嘧啶为中间体,经氯代、缩合、成盐三步反应直接制得盐酸替匹嘧啶,所得原料药无需任何纯化过程,纯度高于99.0%。
附图说明
图1为本发明实施例1所得6-甲酰基尿嘧啶的1H NMR谱图;
图2为本发明实施例2所得6-羟甲基尿嘧啶的1H NMR谱图;
图3为本发明实施例3所得6-氯甲基尿嘧啶的1H NMR谱图;
图4为本发明实施例4所得盐酸替匹嘧啶的1H NMR谱图;
图5为本发明实施例4所得盐酸替匹嘧啶的13C NMR谱图;
图6为本发明实施例4所得盐酸替匹嘧啶的红外谱图;
图7为本发明实施例4所得盐酸替匹嘧啶的HPLC谱图。
具体实施方式
下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供了一种6-氯甲基尿嘧啶的合成方法,包括以下步骤:
将6-甲基尿嘧啶与氧化铜在溶剂中进行氧化反应,得到6-甲酰基尿嘧啶;
还原6-甲酰基尿嘧啶,得到6-羟甲基尿嘧啶;
将6-羟甲基尿嘧啶经氯代反应,得到6-氯甲基尿嘧啶。
本申请提供的6-氯甲基尿嘧啶的合成方法具有环境友好性,且产品收率及纯度高,更适于医药工业应用。
本申请实施例将6-甲基尿嘧啶、溶剂和氧化铜在反应釜中混合,常压向体系中通入空气,加热进行氧化反应,得到6-甲酰基尿嘧啶。
本申请以6-甲基尿嘧啶为原料,以氧化铜为氧化剂,实现了6-甲基尿嘧啶中甲基氧化成甲酰基,制备得到6-甲酰基尿嘧啶。本发明以低毒性的金属氧化剂CuO代替了二氧化硒,实现6-甲基尿嘧啶的氧化工艺,不但能增加环境友好性,而且能提高6-氯甲基尿嘧啶的收率及纯度。
在本发明的实施例中,所述氧化铜与6-甲基尿嘧啶的摩尔比例为1:60~70,优选为1:62~68,更优选为1:63~65。所述氧化反应优选在溶剂乙酸中进行;即本发明实施例可在反应釜中,将6-甲基尿嘧啶加入乙酸中,搅拌下加入氧化铜,加热至一定温度,进行氧化反应。
在本发明的实施例中,所述氧化反应的温度为110~115℃;所述氧化反应的时间为5h~8h。本发明实施例所得反应液的颜色由白色浑浊变为绿色浑浊液,继续保温反应5h~8h,TLC跟踪原料点反应消失,反应完成。其中,TLC采用二氯甲烷:甲醇(体积比)=5:1。
反应完毕后,本发明实施例减压蒸干溶剂,优选将氧化反应得到的混合物在温度为55~65℃且压力为0.09MPa的条件下,减压蒸馏。然后,本发明实施例用硅藻土过滤,得到含6-甲酰基尿嘧啶的滤液。
所述滤液冷却至室温,本发明实施例用酸调节pH值,优选用浓盐酸调节pH值为1~2,可静置8小时~10小时。本发明实施例将所述滤液调节pH值后再过滤,取滤饼醇洗,干燥,得到固体6-甲酰基尿嘧啶。其中,所述过滤可采用本领域常用的抽滤。抽滤得到的滤饼可用10L乙醇洗涤,经60℃干燥7小时~8小时,得到淡黄色固体化合物6-甲酰基尿嘧啶。
采用氧化铜氧化法合成得到6-甲酰基尿嘧啶之后,本申请实施例将其进行还原反应,合成得到6-羟甲基尿嘧啶。在本申请的实施例中,采用硼氢化钠在水存在的条件下还原6-甲酰基尿嘧啶,具体为:
在反应釜中,将6-甲酰基尿嘧啶加入到溶剂如四氢呋喃(THF)中,开启搅拌,加入硼氢化钠(NaBH4),室温搅拌,加入水(H2O)进行还原反应,得到6-羟甲基尿嘧啶。
在本申请的实施例中,所述硼氢化钠与6-甲酰基尿嘧啶的摩尔比例为1:0.8~1.2。所述还原反应的温度可为室温10~30℃;时间为3小时~5小时,TLC跟踪6-甲酰基尿嘧啶点反应消失,反应完成。其中,TLC采用二氯甲烷:甲醇(体积比)=5:1。
本发明实施例将还原反应得到的反应液加入水,减压蒸除溶剂;反应液澄清,冷却至室温,用酸调节pH值,优选用浓盐酸调节pH值为1~2,可静置3小时~4小时,离心,得滤饼和滤液。本发明实施例将滤液减压蒸干,得到固体,再加入水搅拌,抽滤,滤饼合并,干燥,得到类白色固体化合物6-羟甲基尿嘧啶。其中,所述干燥优选为:在60℃减压干燥8小时~10小时。
合成得到6-羟甲基尿嘧啶后,本申请实施例将其进行氯代反应,得到6-氯甲基尿嘧啶。在本申请的实施例中,在二甲基甲酰胺存在的条件下,6-羟甲基尿嘧啶与二氯亚砜进行氯代反应,具体为:
在反应釜中,将6-羟甲基尿嘧啶加入到溶剂如二氯甲烷(DCM)中,开启搅拌,室温下加入二氯亚砜(SOCl2),保持室温加入二甲基甲酰胺(DMF),进行氯代反应,溶液变澄清,反应一定时间,得到6-氯甲基尿嘧啶。
在本申请的实施例中,所述6-羟甲基尿嘧啶与二氯亚砜的摩尔比例为1:10~25。所述氯代反应的温度可为室温10~30℃;时间为3小时~5小时,有大量白色固体析出,TLC跟踪6-羟甲基尿嘧啶点反应消失,反应完成。其中,TLC采用二氯甲烷:甲醇(体积比)=5:1。
本发明实施例将氯代反应得到的反应液离心,取滤饼于60℃鼓风干燥6小时~8小时,得到类白色固体6-氯甲基尿嘧啶。
本发明一些实施例合成6-氯甲基尿嘧啶的反应式如式3所示:
综上所述,本发明以低毒性的金属氧化剂氧化铜代替了二氧化硒,实现了6-甲基尿嘧啶的氧化工艺,进而优化了盐酸替匹嘧啶中间体(6-氯甲基尿嘧啶)的合成方法;所得6-氯甲基尿嘧啶的纯度>99.0%。本发明在增加环境友好性的同时,提高了6-氯甲基尿嘧啶的收率及纯度,利于制备盐酸替匹嘧啶等。
本发明提供了一种盐酸替匹嘧啶的合成方法,包括以下步骤:
将6-甲基尿嘧啶与氧化铜在溶剂中进行氧化反应,得到6-甲酰基尿嘧啶;
还原6-甲酰基尿嘧啶,得到6-羟甲基尿嘧啶;
将6-羟甲基尿嘧啶经氯代反应,得到6-氯甲基尿嘧啶;
将6-氯甲基尿嘧啶依次经过氯代、缩合、成盐,得到盐酸替匹嘧啶。
本发明提供的盐酸替匹嘧啶的合成方法能增加环境友好性,收率高,所得产品纯度高,更安全。
本申请实施例将6-甲基尿嘧啶、溶剂和氧化铜在反应釜中混合,常压向体系中通入空气,加热进行氧化反应,得到6-甲酰基尿嘧啶。
本发明采用6-甲基尿嘧啶,以氧化铜为氧化剂,实现了6-甲基尿嘧啶中甲基氧化成甲酰基,制备得到6-甲酰基尿嘧啶;具体操作步骤等内容如前文所述。本发明以低毒性的金属氧化剂CuO代替了二氧化硒,实现6-甲基尿嘧啶的氧化工艺,在增加环境友好性的同时,提高6-氯甲基尿嘧啶的收率及纯度,进而提高盐酸替匹嘧啶的收率和纯度。
得到6-甲酰基尿嘧啶后,本发明实施例经过还原、氯代反应,制备得到6-氯甲基尿嘧啶。在本发明中,所述还原、氯代反应的内容如前文所述,在此不再一一赘述。
得到6-氯甲基尿嘧啶后,本发明实施例将其进行氯代反应,得到5-氯-6-氯甲基尿嘧啶。
本发明以上述工艺所得6-氯甲基尿嘧啶为中间体,优选经过磺酰氯氯代,具体为:在反应瓶中,将6-氯甲基尿嘧啶加入到溶剂如乙酸中,开启搅拌,冰浴,加入磺酰氯(SO2Cl2),加毕在室温下搅拌反应,得到含5-氯-6-氯甲基尿嘧啶的反应液。
其中,所述6-氯甲基尿嘧啶与磺酰氯的用量比例为1:0.8~1.2(质量g/体积mL)。所述反应优选在室温10~30℃、搅拌的条件下进行;搅拌反应6小时~8小时,有大量白色固体析出。TLC跟踪6-氯甲基尿嘧啶点反应消失(二氯甲烷:甲醇(体积比)=10:1),反应完成。反应液抽滤,所得固体可在60℃鼓风干燥12小时~14小时,得到固体5-氯-6-氯甲基尿嘧啶。
得到5-氯-6-氯甲基尿嘧啶后,本发明实施例优选再与2-氨基吡咯烷盐酸盐缩合,得到替匹嘧啶。
在本发明的实施例中,将5-氯-6-氯甲基尿嘧啶加入到DMF中,开启搅拌,室温下加入2-氨基吡咯烷盐酸盐,水浴下维持体系温度,优选分批加入乙醇钠,搅拌下进行缩合反应,得到替匹嘧啶。
其中,所述5-氯-6-氯甲基尿嘧啶与2-氨基吡咯烷盐酸盐的摩尔比例为1:2~2.5。所述缩合反应的温度可为室温10~30℃;搅拌反应10小时~20小时,有大量白色固体。TLC跟踪5-氯-6-氯甲基尿嘧啶点反应消失(二氯甲烷:甲醇(体积比)=10:1),反应完成。本发明实施例优选冰浴,控制温度30℃以下,向缩合得到的反应液中加入水,搅拌,抽滤。所得固体可在60℃鼓风干燥6小时~8小时,得到类白色固体替匹嘧啶粗品。
在反应瓶中,本发明实施例将替匹嘧啶粗品加入乙醇中,开启搅拌,加入水和盐酸(HCl),加热至回流,进行成盐反应,得到盐酸替匹嘧啶。
在本发明的实施例中,所述替匹嘧啶与盐酸的摩尔比例为1:1~1.5。加热至回流的温度优选为75~80℃,固体全部溶解。本发明实施例趁热过滤,取滤液室温搅拌析晶,抽滤,干燥,得到白色固体盐酸替匹嘧啶精制品。其中,所述室温一般为10~30℃。滤液室温搅拌析晶10小时~20小时;抽滤所得固体可在60℃鼓风干燥12小时~14小时,得到盐酸替匹嘧啶精制品。
基于制备6-氯甲基尿嘧啶所述优化内容,本发明采用所得盐酸替匹嘧啶中间体(6-氯甲基尿嘧啶),可直接制备高纯度的盐酸替匹嘧啶,所得原料药无需任何纯化过程,纯度高于99.0%。
为了进一步理解本申请,下面结合实施例对本申请提供的盐酸替匹嘧啶及其中间体6-氯甲基嘧啶的合成方法进行具体地描述。
实施例1
采用氧化铜氧化法合成6-甲酰基尿嘧啶
在500L反应釜中,将6-甲基尿嘧啶(20.0kg,158.59mol)加入150L乙酸中,搅拌下加入200g氧化铜,常压向体系中通入空气。加热至110℃,反应液颜色由白色浑浊变为绿色混浊液,继续保温反应5h,TLC跟踪原料点反应消失(二氯甲烷:甲醇=5:1),反应完成。
反应完毕后减压(55℃,0.09MPa)蒸干溶剂乙酸,硅藻土过滤,滤液冷却至室温,用浓盐酸调节pH≈1,静置8小时,抽滤,滤饼用10L乙醇洗,60℃鼓风干燥7小时,得淡黄色固体化合物1(8.2kg,收率为37%)。
所得淡黄色固体化合物1的1H NMR结果见图1,图1为本发明实施例1所得6-甲酰基尿嘧啶的1H NMR谱图。结果显示,所得淡黄色固体化合物1为6-甲酰基尿嘧啶。
对比例1
采用二氧化锰氧化法合成6-甲酰基尿嘧啶
在5L反应釜中,将6-甲基尿嘧啶(200.0g,1.59mol)加入1.5L乙酸中,搅拌下加入2.0g二氧化锰,常压向体系中通入空气。加热至110~115℃,反应液颜色由白色浑浊变为绿色混浊液,继续保温反应24h,TLC跟踪原料点大量剩余(二氯甲烷:甲醇=5:1),采用柱层析分离产品(二氯甲烷:甲醇=20:1)。
含有产物的馏分合并后浓缩,得淡黄色固体化合物1(2.1g,收率为10%)。
对比例2
采用二氧化硒氧化法合成6-甲酰基尿嘧啶
在5L反应釜中,将6-甲基尿嘧啶(200.0g,1.59mol)加入1.5L乙酸中,搅拌下加入2.0g二氧化硒,常压向体系中通入空气。加热至110~115℃,反应液颜色由白色浑浊变为绿色混浊液,继续保温反应5h,TLC跟踪原料点反应消失(二氯甲烷:甲醇=5:1),反应完成。
反应完毕后减压(55℃,0.09MPa)蒸干溶剂乙酸,硅藻土过滤,滤液冷却至室温,用浓盐酸调节pH≈1,静置8小时,抽滤,滤饼用100mL乙醇洗,60℃鼓风干燥7小时,得淡黄色固体化合物1(4.2g,收率为20%)。
由以上内容可知,按照本发明所述制备方法,所得6-氯甲基尿嘧啶的纯度>99.0%。本发明优化了盐酸替匹嘧啶中间体(6-氯甲基尿嘧啶)的合成方法,以低毒性的金属氧化剂氧化铜代替了二氧化硒,实现了6-甲基尿嘧啶的氧化工艺,增加环境友好性,同时提高了6-氯甲基尿嘧啶的收率及纯度,这对于医药工业应用具有重要的实践意义。
实施例2
6-羟甲基尿嘧啶的合成
在500L反应釜中,将化合物1(8.2kg,58.53mol)加入到82L四氢呋喃中,开启搅拌,加入硼氢化钠(2.38kg,64.38mol),室温(20℃)搅拌20min,慢慢滴加100mL水,有气泡产生,保持温度(20℃)搅拌3小时,TLC跟踪化合物1点反应消失(二氯甲烷:甲醇=5:1),反应完成。
反应液慢慢加入42L水,减压蒸除四氢呋喃,反应液澄清,冷却至室温,用浓盐酸调节pH≈1,静置3小时,离心,得滤饼;滤液减压蒸干得固体,加入10L水搅拌30min,抽滤,滤饼合并,60℃减压干燥8小时,得类白色固体化合物2(8.0kg,收率为96.20%)。
所得类白色固体化合物2的1H NMR结果见图2,图2为本发明实施例2所得6-羟甲基尿嘧啶的1H NMR谱图。结果显示,所得类白色固体化合物2为6-羟甲基尿嘧啶,纯度为98.8%。
实施例3
6-氯甲基尿嘧啶的合成
在500L反应釜中,将化合物2(8.0kg,56.29mol)加入到80L二氯甲烷中,开启搅拌,室温下(20℃)加入8L二氯亚砜,保持室温慢慢滴加4L DMF,溶液变澄清,反应3h,有大量白色固体析出。TLC跟踪化合物2点反应消失(二氯甲烷:甲醇=5:1),反应完成。反应液离心,滤饼60℃鼓风干燥6小时,得类白色固体SM1(5.0kg,收率为55.56%)。
所得类白色固体SM1的1H NMR结果见图3,图3为本发明实施例3所得6-氯甲基尿嘧啶的1H NMR谱图。结果显示,所得类白色固体SM1为6-氯甲基尿嘧啶,纯度为99.2%。
实施例4
盐酸替匹嘧啶的合成
在20L反应瓶中,将化合物SM1(1000.0g,6.23mol)加入到5000mL乙酸中,开启机械搅拌,冰浴,滴加磺酰氯(1000mL,12.37mol),保持滴加过程温度不超过30℃,加毕在室温下(20℃)搅拌反应6h,有大量白色固体析出。TLC跟踪化合物SM1点反应消失(二氯甲烷:甲醇=10:1),反应完成。反应液抽滤,所得固体在60℃鼓风干燥12小时,得5-氯-6-氯甲基尿嘧啶。
在20L反应瓶中,将5-氯-6-氯甲基尿嘧啶(950.0g,4.87mol)加入到9500mLDMF中,开启机械搅拌,室温(20℃)下加入2-氨基吡咯烷盐酸盐(1175.0g,9.74mol),水浴下维持体系温度为20℃,分批加入乙醇钠(994.6g,14.61mol),搅拌反应10h,有大量白色固体。TLC跟踪5-氯-6-氯甲基尿嘧啶点反应消失(二氯甲烷:甲醇=10:1),反应完成。冰浴,控制温度30℃以下,向反应液中加入9500mL水,搅拌30min,抽滤。所得固体60℃鼓风干燥6小时,得类白色固体替匹嘧啶粗品685.6g。
在20L反应瓶中,将替匹嘧啶粗品(680.0g,2.80mol)加入到6800mL乙醇中,开启机械搅拌,加入3400mL水,1M HCl 2000mL,加热至回流(75℃),固体全部溶解,趁热过滤,滤液室温(20℃)搅拌析晶10小时,抽滤,所得固体60℃鼓风干燥12小时,得白色固体盐酸替匹嘧啶精制品475.5g。
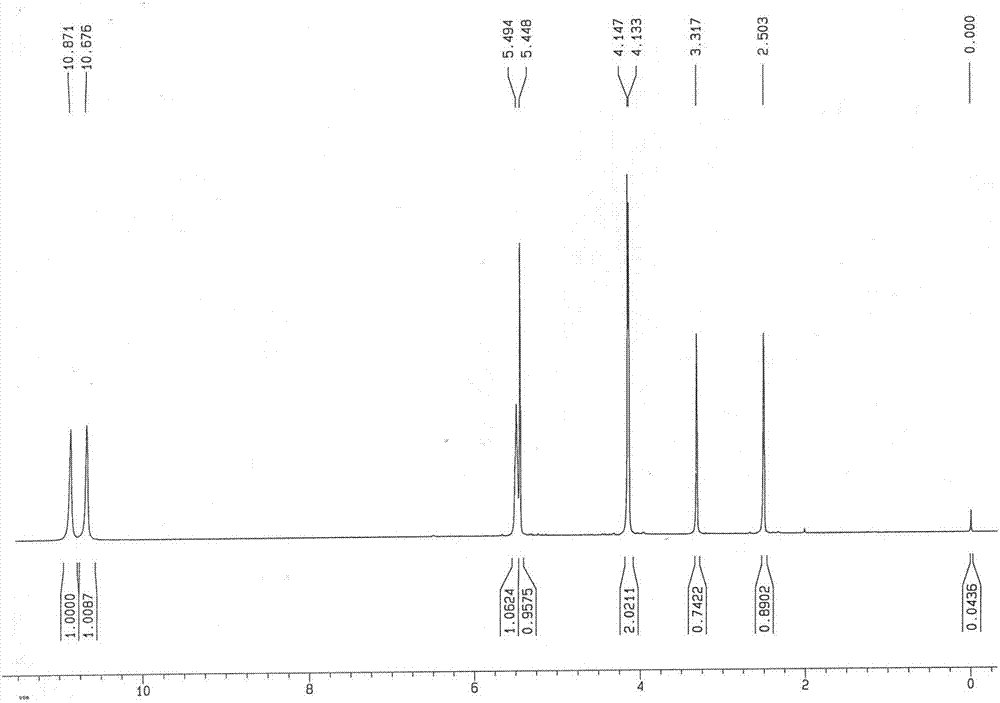
通过核磁共振分析和红外分析,对所得白色固体盐酸替匹嘧啶精制品进行结构鉴定,1H NMR结果见图4,图4为本发明实施例4所得盐酸替匹嘧啶的1H NMR谱图;13C NMR结果见图5,图5为本发明实施例4所得盐酸替匹嘧啶的13C NMR谱图;红外结果见图6,图6为本发明实施例4所得盐酸替匹嘧啶的红外谱图。
采用本领域常规的高效液相色谱法,对所得白色固体盐酸替匹嘧啶精制品进行分析。其中,进样次数为1,进样体积为10μL,运行时间为40min;采集方法组为TPI有关物质方法,处理方法为TPI处理方法;通道名称为2998Ch1 210nm@1.2nm。图7为本发明实施例4所得盐酸替匹嘧啶的HPLC谱图,表1为色谱峰结果。本发明所得盐酸替匹嘧啶纯度>99%,单杂<0.2%(见图7,产品纯度为99.959%)。
表1色谱峰结果
由以上实施例可知,本发明采用上述工艺所得6-氯甲基尿嘧啶为中间体,经氯代、缩合、成盐三步反应直接制得盐酸替匹嘧啶,所得原料药无需任何纯化过程,纯度高于99.0%。
以上所述仅是本发明的优选实施方式,应当指出,对于使本技术领域的专业技术人员,在不脱离本发明技术原理的前提下,是能够实现对这些实施例的多种修改的,而这些修改也应视为本发明应该保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!