一种螺芴化合物及其发光器件的制作方法
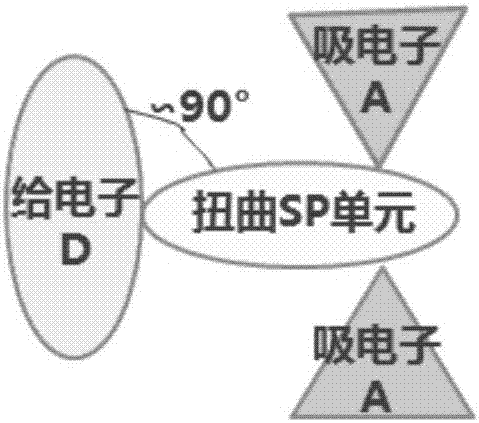
本发明涉及有机发光材料
技术领域:
,具体涉及一种螺芴化合物及其发光器件。
背景技术:
:根据电致发光机制,oled材料分为荧光oled材料和磷光oled。现有oled技术材料,磷光发光材料由于含有重金属效应,理论上可以达到100%的量子发光效率,尤其在红光和绿光磷光材料方面已经取得了长足的发展。但由于磷光材料的三线态激子在高浓度下容易发生猝灭,因此,为提高性能,需要保持一定的主体客体掺杂比例。荧光oled材料是一种纯粹的有机材料,不含有重金属,因此,理论上只能达到25%的内部量子效率,造成理论上荧光的外部量子效率最高5%的上限。目前红光和绿光oled材料已经取得了长足的发展,荧光蓝oled材料在性能上尚未能跟其它红绿光材料进行媲美。近期,热激活延迟荧光(thermallyactivateddelayedfluorescence,tadf)材料得到广泛关注,这种材料由于homo-lumo轨道分离,使得三线态激子可以通过热的方式跳跃到单线态轨道上,因而可以获得接近100%的内部量子效率。然而,蓝光tadf材料依然是目前的一个难点,这是由于要获得色纯度高的荧光蓝,至少要求有机材料的三线态达到2.6ev以上,并保持较高性能。世界范围内,很多科研院所致力于开发这类材料,但成果不多。鉴于此,特提出本发明。技术实现要素:本发明的第一目的在于提供一种作为荧光蓝材料的螺二芴化合物。本发明的第二目的在于提供使用该螺二芴化合物的发光器件。为了完成本发明的目的,采用的技术方案为:为了实现本发明的第一目的,本发明提出一种螺二芴化合物,选自如通式所示i的化合物:其中,y1、y2各自独立的表示氢、吸电子基团或供电子基团;x1、x2中至少有一个取代基为式ii所示的取代基:m表示-s-、-p-、-so-、-so2-、-s(=s)-、-s(=s)(=s)-、-po-、-po2-、-p(=s)-、-p(=s)(=s)-、-c(=o)-;n1、n2、n3、n4各自独立的表示碳原子或氮原子;ra选自为氢、卤素、c1~30烷基、羟基取代的c1~30烷基或c6~48烷基芳基;n为0~4的整数。为了实现本发明的第二目的,所述发光器件包括阳极、阴极及设置于所述阳极和所述阴极之间的至少一个有机层,所述有机层包括本发明所述的螺芴化合物。本发明的技术方案至少具有以下有益的效果:本发明提出一种基于螺二芴结构的热激活延迟荧光材料,具有a-d-a化学结构,其中,给电子d单元与吸电子a单元之间形成接近90°的空间二面角,有利于热激活延迟荧光材料的homo-lumo轨道分离,以获得理想的δest和其它光电性质。同时,本发明的螺二芴化合物具有很宽的homo-lumo能带,非常低的homo值,也非常适合于hbl或host材料。附图说明图1为本发明发光器件的结构示意图;图2为本发明螺二芴化合物的a-d-a空间构型示意图;图3为本发明螺二芴化合物的又一a-d-a空间构型示意图;图4为化合物sdf-dpso2的紫外吸收光谱;图5为化合物sdf-dpso2的核磁共振碳谱;图6为化合物sdf-dpso2的核磁共振氢谱;图7为化合物sdf-dpso2的分子基态模拟图;图8为化合物sdf-dpyso2的核磁共振碳谱;图9为化合物sdf-dpyso2的核磁共振氢谱;图10为化合物sdf-dpsocl2的核磁共振碳谱;图11为化合物sdf-dpsocl2的核磁共振氢谱;图12为化合物sdf-4pysocl的核磁共振碳谱;图13为化合物sdf-4pysocl的核磁共振氢谱。10-发光器件;11-阳极;12-空穴传输层;13-发光层;14-电子传输层;15-阴极。具体实施方式下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本申请而不用于限制本申请的范围。本发明涉及一种螺二芴化合物,螺二芴化合物选自如通式所示i的化合物:其中,y1、y2各自独立的表示氢、吸电子基团或供电子基团;x1、x2中至少有一个取代基为式ii所示的取代基:m表示-s-、-p-、-so-、-so2-、-s(=s)-、-s(=s)(=s)-、-po-、-po2-、-p(=s)-、-p(=s)(=s)-、-c(=o)-;n1、n2、n3、n4各自独立的表示碳原子或氮原子;ra选自为氢、卤素、c1~30烷基、羟基取代的c1~30烷基或c6~48烷基芳基;n为0~4的整数。本发明提出的螺二芴化合物,具有a-d-a化学结构,其空间构型的示意图如图1和图2所示。根据图1和图2中所示,给电子d单元与吸电子a单元之间形成接近90°的空间二面角,有利于热激活延迟荧光材料的homo-lumo轨道分离。本发明的螺二芴化合物,通过给电子d-sp单元保持高三线态和单线态能级以得到纯蓝光光谱,本发明中电子d单元与吸电子a单元之间形成接近90°空间结构,有利于防止给电子d和sp之间,以及给电子d和吸电子a之间形成共轭,以至于光谱红移。作为本发明螺二芴化合物的一种改进,螺二芴化合物选自如通式所示ia的化合物:在式ia中,x1、x2、y1、y2所在的取代位点是芴的2、7位取代基是芴发生化学活性最高的位点,因此式ia所表示的化合物结构更易于合成。作为本发明螺二芴化合物的一种改进,当y1、y2为供电子基团供电子基团(称为给电子基团)时,y1、y2各自独立的选自取代或未取代的c1~30烷基、取代或未取代的二联苯基、或以下结构式表示的取代基:其中,r1、r2、r3、r4各自独立的选自氢原子、氨基、卤素、取代或未取代的c1~12烷基、取代或未取代的c1~12烷氧基、取代或未取代的c6~12芳基、取代或未取代的c6~12芳氧基;取代基为卤素原子、c1~12的烷基、卤素原子取代的c1~12的烷基、卤素原子取代的c1~12的烷氧基;m为0~4的整数;式y-6、式y-7、式y-11、式y-12、式y-16和式y-17所示的基团中苯环上的任意氢原子可被取代而形成取代基。作为本发明螺二芴化合物的一种改进,当y1、y2为吸电子基团供电子基团(称为拉电子基团)时,y1、y2各自独立的式ii所示的取代基。作为本发明螺二芴化合物的一种改进,在式i中,x1选自氢或式iia所示的取代基,x2选自式iia所示的取代基;k表示碳原子或氮原子。作为本发明螺二芴化合物的一种改进,在式iia中,m表示-so-、-so2-、-po-。作为本发明螺二芴化合物的一种改进,当y1、y2为供电子基团时,螺二芴化合物选自如通式ia-1所示的化合物:在ia-1中,m1、m2各自独立的表示-so-、-so2-、-po-;ra1、ra2各自独立的选自氢、卤素、c1~12烷基、c6~24芳基;k1、k2各自独立的表示碳原子或氮原子。y1、y2各自独立的选自本发明中的供电子基团。进一步优选的,ra1、ra2各自独立的选自氢原子或卤素。更优选的,m1、m2相同;ra1、ra2相同;k1、k2相同。如果y1、y2为供电子取代基,即在homo轨道的离域位置加上供电子取代基,届时homo轨道的分布会更加分散到这些供电子取代基上,但不会影响到lomo,材料保持tadf特性。作为本发明螺二芴化合物的一种改进,当y1、y2为氢原子时,螺二芴化合物选自如通式ia-2所示的化合物:在ia-2中,m1、m2各自独立的表示-so-、-so2-、-po-;ra1、ra2各自独立的选自氢、卤素、c1~12烷基、c6~24芳基;k1、k2各自独立的表示碳原子或氮原子。进一步优选的,ra1、ra2各自独立的选自氢原子或卤素。更优选的,m1、m2相同;ra1、ra2相同;k1、k2相同。作为本发明螺二芴化合物的一种改进,当y1、y2为吸电子基团时,螺二芴化合物选自如通式ia-3所示的化合物:在ia-3中,m1、m2、m3、m4各自独立的表示-so-、-so2-、-po-;ra1、ra2、ra3、ra4各自独立的选自氢、卤素、c1~12烷基、c6~24芳基;k1、k2、k3、k4各自独立的表示碳原子或氮原子;n1、n2、n3、n4各自独立的选自0~4的整数。进一步优选的,ra1、ra2、ra3、ra4各自独立的选自氢原子或卤素。更优选的,m1、m2、m3、m4相同;ra1、ra2、ra3、ra4相同;k1、k2、k3、k4相同。如果y1、y2为吸电子取代基,lumo轨道更加离域,更加分散,与homo轨道的重叠成分更少。作为本发明螺二芴化合物的一种改进,当y1、y2为氢原子时,螺二芴化合物选自以下结构式所示的化合物:作为本发明螺二芴化合物的一种改进,当y1、y2为供电子基团时,螺二芴化合物选自以下结构式所示的化合物:作为本发明螺二芴化合物的一种改进,当y1、y2为吸电子基团时,螺二芴化合物选自以下结构式所示的化合物:螺二芴化合物的合成路线为:具体工艺为:在装有a的反应瓶中滴加nbs(n-溴代琥珀酰亚胺)/thf的混合溶液,在氮气保护条件下加热到40℃保温1小时,进行溴代反应得到b。然后,加入双取代的二硫化合物r-s-s-r,以n-buli/thf作为催化剂,在低温干冰浴中充分搅拌半个小时,得到化合物c。最后,通入间位氯代苯甲酸mcpba/ch2cl2混合溶液,进行充分搅拌1小时,加入水,析出固体,依次使用正己烷洗涤,乙醇进行重结晶,得到d。r可以是苯环,吡啶,对位氯苯,间位氯代吡啶。r’=h或d=r’=h或d=r’=h或d=r’=h或d=r’=h或d=r’=h或d=r’=h或则d=r’=h或则d=下面继续举例说明本发明材料的合成:合成实施例1:化合物sdf-dpso2的合成在装有1mola的反应瓶中滴加nbs(n-溴代琥珀酰亚胺)/thf的混合溶液,在氮气保护条件下加热到40c保温1小时,进行溴代反应得到b。然后,加入双苯取代的二硫化合物ph-s-s-ph,以n-buli/thf作为催化剂,在低温干冰浴中充分搅拌半个小时,得到化合物c。最后,通入间位氯代苯甲酸mcpba/ch2cl2混合溶液,进行充分搅拌1小时,加入水,析出固体,依次使用正己烷洗涤,乙醇进行重结晶,得到sdf-dpso2,产率38%。其紫外吸收光谱(ch2cl2)如图4所示,根据紫外吸收光谱可知,该化合物在250~400nm之间有比较强的吸收光谱,其中,360nm和310nm处的吸收强度最高。其光致发光光谱(pl光谱)的主峰在435.22nm,是一种蓝光材料。其核磁共振碳谱如图5所示、核磁氢谱如图6所示。对采用gaussian03量化模拟软件对sdf-dpso2分子基态结构进行模拟,其分子基态模拟图如图7所示,模拟图如图7所示;由图7可知,homo分布在螺旋化合物上,lumo分布在两侧的苯基亚砜上,预期可以达到比较低的δest单线态与三线态分裂能,homo-lumo轨道分离比较彻底。采用tddft(含时密度泛函)方法,在b3lyp水平下进行模拟材料基态时的分子构型,计算其键长和键角,具体如表1所示:表1键长a键角二面角c7-c81.46943c1-c21.53385c1-c131.53336c1-c141.53053c1-c251.53053s1-c161.80379s1-c321.80352s2-c231.80379s2-c261.80352c2-c1-c13101.425c14-c1-c25101.086c32-s1-c16104.771c23-s2-c26104.770c13-c1-c2-c14122.506根据表1分子数据可以看出,sdf-dpso2化合物保持比较好的分子对称性,二面角c13-c1-c2-c14=122°,使得homo-lumo分离。合成实施例2:化合物sdf-dpyso2的合成在装有1mola的反应瓶中滴加br2/ch3cooh的混合溶液,在氮气保护条件下进行溴代反应得到b。然后,加入双吡啶取代的二硫化合物py-s-s-py,以n-buli/thf作为催化剂,在低温干冰浴中充分搅拌半个小时,得到化合物c。最后,通入间位氯代苯甲酸mcpba/ch2cl2混合溶液,进行充分搅拌1小时,加入水,析出固体,依次使用正己烷洗涤,乙醇进行重结晶,得到sdf-dpyso2,产率46%。其核磁共振碳谱如图8所示、核磁氢谱如图9所示。合成实施例3:化合物sdf-dpysocl2的合成在装有1mola的反应瓶中滴加br2/ch3cooh的混合溶液,在氮气保护条件下进行溴代反应得到b。然后,加入双吡啶取代的二硫化合物pycl-s-s-pycl,以n-buli/thf作为催化剂,在低温干冰浴中充分搅拌半个小时,得到化合物c。最后,通入间位氯代苯甲酸mcpba/ch2cl2混合溶液,进行充分搅拌1小时,加入水,析出固体,依次使用正己烷洗涤,乙醇进行重结晶,得到sdf-dpysocl2,产率43%。其核磁共振碳谱如图10所示、核磁氢谱如图11所示。合成实施例4:化合物sdf-4pysocl的合成在装有a的反应瓶中滴加br2/ch3cooh的混合溶液,在氮气保护条件下进行溴代反应得到b。然后,加入双氯代吡啶取代的二硫化合物pycl-s-s-pycl,以n-buli/thf作为催化剂,在低温干冰浴中充分搅拌半个小时,得到化合物c。最后,通入间位氯代苯甲酸mcpba/ch2cl2混合溶液,进行充分搅拌1小时,加入水,析出固体,依次使用正己烷洗涤,乙醇进行重结晶,得到sdf-4pysocl,产率63%。其核磁共振碳谱如图12所示、核磁氢谱如图13所示。δest测试一般有机材料中,由于自旋度不同而造成s1激发态和t1激发态能量不同,且es1能量要比et1能量大0.5-1.0ev,这就造成纯有机荧光材料发光效率低下。热延迟荧光tadf材料,由于独特分子设计,将homo-lumo轨道进行分离,降低二者电子交换能,理论上可以实现δest∽0。为了有效评估本发明中材料的热延迟荧光效果,进行δest评估。将1wt%化合物掺杂到mcbp薄膜中,在77k条件下进行荧光发射光谱和磷光发射光谱量测,通过波长与能量关系进行换算成s1和t1值(e=1240/λem)。然后,δest=es1-et1获得单线体和三线态的分裂能。数据如表2所示:表2:测试项目sdf-dpso2sdf-dpyso2sdf-dpsocl2sdf-4pysocles1(ev)2.752.762.742.81et1(ev)2.612.642.632.72δest(ev)0.140.120.110.09从表2可知,本发明各个化合物都具有比较小的δest值,都小于0.2ev,因此,都具备热延迟荧光效果。本发明还涉及一种发光器件,其发光器件为有机发光二极管(oled)。包括阳极、阴极及设置于阳极和阴极之间的至少一个有机层,有机层包括本发明的芳香族化合物。请参阅图1,为本发明提供的发光器件的结构示意图。发光器件10包括依次沉积形成的阳极11、空穴传输层12、发光层13、电子传输层14和阴极15。其中空穴传输层12、发光层13、电子传输层14均为有机层,阳极11与阴极15与电连接。ito基板是30mm×30mm尺寸的底发射玻璃,有四个发光区域,发光面积aa区为2mm×2mm,ito薄膜的透光率为90%@550nm,表面粗糙度ra<1nm,ito膜厚为1300a,方电阻为10欧姆每平方。ito基板的清洗方式是,首先放置在盛有丙酮溶液的容器中,将该容器放置于在超声波清洗机进行超声清洗,清洗时间为30分钟,主要是将附着在ito表面的有机物进行溶解和祛除;然后将清洗完毕的ito基板取出放置在热板上进行高温120烘烤半个小时,主要是移除ito基板表面的有机溶剂和水汽;然后将烘烤完毕的ito基板迅速转移到uv-zone设备中进行o3plasma处理,将ito表面难以除尽的有机物或异物进一步使用等离子处理,处理时间为15分钟,处理完毕的ito要迅速转移到oled蒸镀设备成膜室中。oled蒸镀前准备:首先对oled蒸镀设备进行洁净处理,使用ipa进行擦拭成膜室的腔体内壁,保证整个成膜腔体没有异物或粉尘。然后,将装有oled有机材料的坩埚和装有金属铝粒的坩埚依次放置在有机蒸发源和无机蒸发源位置上。关闭腔体,进行初抽真空和抽高真空步骤,使得oled蒸镀设备内部蒸镀度达到10-7torr。oled蒸镀成膜:打开oled有机蒸发源,对oled有机材料进行100c预热,预热时间为15分钟,保证进一步移除oled有机材料中的水汽。然后对需要蒸镀的有机材料进行快速升温加热处理,并打开蒸发源上方的挡板,直到该材料的蒸发源有有机材料跑出,同时晶振片检测器检测到蒸发速率时,然后进行缓慢升温,升温幅度为1~5℃,直到蒸发速率稳定在1a/秒时,打开掩膜板板正下方的挡板,进行oled成膜,当电脑端观测到ito基板上的有机膜达到预设膜厚时,关闭掩膜板挡板和蒸发源正上方挡板,关闭该有机材料的蒸发源加热器。其它有机材料和阴极金属材料的蒸镀工艺如上所述。oled封装流程:20mm×20mm的封装盖的清洁处理方式如ito基板前处理方式。在清洁完毕的封装盖外延四周进行uv胶材涂覆或点胶,然后,将点完uv胶材的封装盖转移到真空贴合设备中,与成膜oled有机膜的ito基板进行真空贴合,然后,转移到uv固化腔体中,使用365nm波段的紫外光进行光固化。光固化的ito器件,还需要进行80℃半小时的后热处理,使得uv胶材完全固化。(1)作为客体发光材料性能评估为了评估sdf-dpso2作为客体发光材料的电致发光性能,设计oled器件结构ito/npb(30nm)/tcta(30nm)/ppf:sdf-dpso2(xwt%,30nm,x=1-20)/ppf(10nm)/tpbi(30nm)/lif(0.8nm)/al(150nm)。封装后的样品进行ivl性能测试,ivl设备采用mcsciencem6100进行测试,测试数据如表3所示:表3采用经典的磷光蓝firpic材料进行性能对比(编号f),设计了oled器件结构ito/npb(30nm)/tcta(30nm)/ppf:sdf-dpso2(10wt%,30nm)/ppf(10nm)/tpbi(30nm)/lif(0.8nm)/al(150nm)。可以发现,基于sdf-dpso2的器件性能随着掺杂比例提高而提高,掺杂比例继续提高后,器件性能有下降的趋势,其中,掺杂比例为10wt%的sdf-dpso2性能已经非常接近磷光蓝材料的性能。(2)作为主体材料的光电性能评估:器件制作过程如上所述。设计oled器件结构(编号g),oled器件结构ito/npb/tcta/sdf-dpso2:firpic(10wt%/sdf-dpso2(10nm)/tpbi(30nm)/lif(0.8nm)/al,发现g的器件性能要远远好于f,这是因为sdf-pso材料同时具有传输电子和空穴的能力,而且sdf-dpso2材料的homo-lumo大于firpic的homo-lumo,因此能量传递良好。本申请虽然以较佳实施例公开如上,但并不是用来限定权利要求,任何本领域技术人员在不脱离本申请构思的前提下,都可以做出若干可能的变动和修改,因此本申请的保护范围应当以本申请权利要求所界定的范围为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 螺[芴‑9,9‑氧杂蒽]类空穴传输材料及其应用的制造方法与工艺
- 可采用环境友好溶剂加工的以螺芴单元为核的双极性小分子发光材料及其制备方法与应用与流程
- 一种含有氮杂螺芴和含氮六元杂环的化合物及其在OLED上的应用的制造方法与工艺
- 一种含有氮杂螺芴和含氮六元杂环的化合物及其在有机电致发光器件上的应用的制造方法与工艺
- 一种以吖啶螺蒽酮为核心的化合物及其在有机电致发光器件上的应用的制造方法与工艺
- 一种含有吖啶螺蒽酮类化合物的有机电致发光器件及其应用的制造方法与工艺
- 螺双芴衍生物及其在有机电致发光领域中的应用的制造方法与工艺
- 一种螺硅芴衍生物及其有机发光器件的制造方法与工艺
- 螺芴类衍生物及其制备方法和有机发光器件与制造工艺
- 一种4‑溴螺环[芴‑9,9’‑氧杂蒽]的合成方法与制造工艺