两种蒽醌类化合物的制备方法和应用与流程
本发明属于特色民族药用植物中的有效成分提取分离以及活性筛选的技术领域,具体涉及两种蒽醌类化合物及其制备方法和应用。
背景技术:
豆科苏木亚科决明属木本植物腊肠树(cassiafistula),为傣族地区常用药用植物,原产于印度、缅甸和斯里兰卡,在我国南部和西南部均有栽培,是南方常见的庭院观赏树木,树皮含单宁,可做红色染料;根、树皮、果瓤和种子均可入药作缓泻剂;木材坚重,耐朽力强,可作支柱、桥梁、车辆及农具等用材;而其在民间被用于治疗皮肤病、周期性发热和肿瘤等疾病,更是突显出了其一定的药用价值;本发明从腊肠树中分离得到两个新的具有一定的细胞毒活性和抗烟草花叶病毒活性的蒽醌类化合物,其中,fistulaquinonea的6位具有少见的羟乙基取代,结构具有一定的特点。
技术实现要素:
本发明的第一目的是提供两个新的蒽醌类化合物;第二目的在于提供所述蒽醌类化合物的制备方法;第三目的在于提供所述蒽醌类化合物在制备抗癌和抗烟草花叶病毒药物中的应用。
本发明的第一目的是这样实现的,所述的两个蒽醌类化合物是从豆科苏木亚科决明属木本植物腊肠树(cassiafistula)的干燥的细枝中分离得到,分别命名为fistulaquinonea(分子式:c19h18o5)和fistulaquinoneb(分子式:c19h18o5)分别具有下述结构,其中,fistulaquinonea的6位具有少见的羟乙基取代:
本发明的第二目的是这样实现的,所述的蒽醌类化合物的制备方法,其特征在于蒽醌类化合物是以豆科苏木亚科决明属木本植物腊肠树(cassiafistula)的干燥的细枝为原料,经有机溶剂提取、mci脱色、硅胶柱层析、高效液相色谱分离步骤而得到具体为:
a、浸膏提取:将豆科苏木亚科决明属木本植物腊肠树(cassiafistula)的干燥的细枝20~40目粉碎,用有机溶剂超声提取3~5次,每次30~60分钟,提取液合并;提取液过滤,减压浓缩提取液,静置,滤除沉淀物,浓缩成浸膏a;
b、mci脱色:浸膏a,用重量比2~5倍量的甲醇水溶解,上mci柱,用80~90%甲醇水洗脱,合并洗脱液,减压浓缩成浸膏b;
c、硅胶柱层析:将b步骤所得的浸膏b用重量比1~2倍量的甲醇或者丙酮溶解,用浸膏重1~2倍的80~100目硅胶拌样,然后上硅胶柱层析(装柱硅胶为100~200目),用量为浸膏b重量6~10倍量;用体积比为1:0~0:1的混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;
d、将9:1洗脱部分再次用体积比为9:1~1:2混合有机溶剂洗脱;
e、高效液相色谱分离:以50-80%的甲醇为流动相,流速8~12ml/min,21.2´250mm,5mm的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为202~280nm,每次进样10~100ml,收集10~50min的色谱峰,多次累加后蒸干,即得所述的2个蒽醌类化合物;
其中a步骤所用的有机溶剂为70%的丙酮;b步骤所用的有机溶剂为90%甲醇;c步骤所用的混合有机溶剂为氯仿和丙酮的体积比配比为20:1,9:1,8:2,7:3,6:4和1:1;d步骤所用的混合有机溶剂乙醚和丙酮的体积配比为9:1,8:2,7:3,6:4,1:1和1:2。
本发明两个新的恩醌类化合物结构是通过核磁共振和质谱测定方法确定,并表征其具体结构为:
本发明的第三目的是这样实现的,所述的蒽醌类化合物在制备抗癌和抗烟草花叶病毒药物中的应用。
本发明的两个新蒽醌类化合物经对抗烟草花叶病毒的实验,其中fistulaquinonea和fistulaquinoneb的相对抑制率在20μm下分别达到25.2%和26.3%,低于阳性对照品南宁霉素的相对抑制率(28.8%),说明fistulaquinonea和fistulaquinoneb具有一定的抗烟草花叶病毒活性;此外,本发明的两个新蒽醌类化合物对白血病急性早幼粒nb4细胞、肺腺癌a549细胞、人骨髓神经母细胞瘤shsy5y细胞、人前列腺癌pc3细胞、人乳腺癌mcf7细胞的细胞毒活性测试;其中,蒽醌类化合物fistulaquinonea对nb4和shsy5y细胞株具有一定的细胞毒活性,ic50值分别达6.2和8.4μm;蒽醌类化合物fistulaquinoneb对a549细胞株具有一定的细胞毒活性,ic50值为5.5μm。
本发明化合物结构新颖活性好,可作为抗癌和抗烟草花叶病毒药物的先导性化合物,在药物发现方面有良好的应用前景。
附图说明
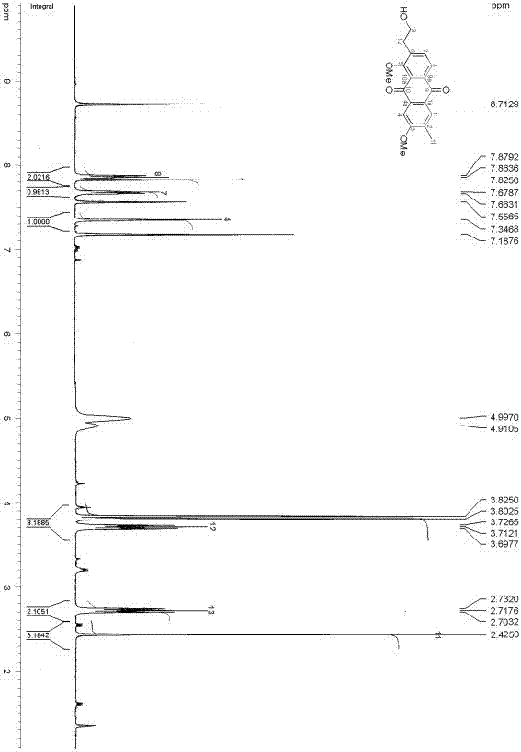
图1为本发明化合物fistulaquinonea的核磁共振碳谱(13cnmr)。
图2为本发明化合物fistulaquinonea的核磁共振氢谱(1hnmr)。
图3为本发明化合物fistulaquinoneb的核磁共振氢谱(13cnmr)。
图4为本发明化合物fistulaquinoneb的核磁共振氢谱(1hnmr)。
图5为化合物fistulaquinonesa和b的1d核磁数据(溶剂为c5d5n)(125and500mhz)。
图6为两个新蒽醌类化合物的细胞毒活性。
具体实施方式
下面结合附图对本发明作进一步的说明,但不以任何方式对本发明加以限制,基于本发明教导所作的任何变换或改进,均落入本发明的保护范围。
本发明所述两个蒽醌类化合物是从豆科苏木亚科决明属木本植物腊肠树(cassiafistula)的干燥的细枝中分离得到,分别命名为fistulaquinonea(分子式:c19h18o5)和fistulaquinoneb(分子式:c19h18o5),分别具有下述结构:
其特征在于蒽醌类化合物是以豆科苏木亚科决明属木本植物腊肠树(cassiafistula)的干燥的细枝为原料,经有机溶剂提取、mci脱色、硅胶柱层析、高效液相色谱分离步骤而得到具体为:
a、浸膏提取:将豆科苏木亚科决明属木本植物腊肠树(cassiafistula)的干燥的细枝20~40目粉碎,用有机溶剂超声提取3~5次,每次30~60分钟,提取液合并;提取液过滤,减压浓缩提取液,静置,滤除沉淀物,浓缩成浸膏a;
b、mci脱色:浸膏a,用重量比2~5倍量的甲醇水溶解,上mci柱,用80~90%甲醇水洗脱,合并洗脱液,减压浓缩成浸膏b;
c、硅胶柱层析:将b步骤所得的浸膏b用重量比1~2倍量的甲醇或者丙酮溶解,用浸膏重1~2倍的80~100目硅胶拌样,然后上硅胶柱层析(装柱硅胶为100~200目),用量为浸膏b重量6~10倍量;用体积比为1:0~0:1的混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;
d、将9:1洗脱部分再次用体积比为9:1~1:2混合有机溶剂洗脱;
e、高效液相色谱分离:以50~80%的甲醇为流动相,流速8~12ml/min,21.2´250mm,5mm的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为202~280nm,每次进样10~100ml,收集10~50min的色谱峰,多次累加后蒸干,即得所述的2个蒽醌类化合物;
其中a步骤所用的有机溶剂为70%的丙酮;b步骤所用的有机溶剂为90%甲醇;c步骤所用的混合有机溶剂为氯仿和丙酮的体积比配比为20:1,9:1,8:2,7:3,6:4和1:1;d步骤所用的混合有机溶剂乙醚和丙酮的体积配比为9:1,8:2,7:3,6:4,1:1和1:2。
本发明所述的腊肠树植物不受地区和品种限制,均可以实现本发明。
实施例1
取干燥的豆科苏木亚科决明属木本植物腊肠树(cassiafistula)的细枝2.2kg,粗粉碎至40目,用70%的丙酮超声提取4次,每次60分钟,提取液合并;提取液过滤,减压浓缩至体积的1/4;静置,滤除沉淀物,浓缩成63.5g的浸膏a;浸膏a用mci装柱,在浸膏a中加入150g的80%甲醇水溶解,用90%甲醇水2~4l洗脱,收集洗脱液,减压浓缩得到31.2g浸膏c,浸膏c中加60g丙酮,然后加入100目硅胶60g拌样,拌样后,用200目硅胶500g装柱;用体积比分别为0:1,9:1,8:2,7:3,6:4和1:1的氯仿-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分,得到6个部分a-f,其中,对收集到的样品2部分6.42g,再重复硅胶柱层析,用体积比分别为9:1,8:2,7:3,6:4,1:1和1:2的乙醚-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分,得到6个部分b1-b6,其中第b3部分,即7:3部分约650mg,再以50~80%的甲醇为流动相,流速12ml/min,21.2´250mm,5mm的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为254nm,每次进样50ml,分别收集15min及21min的色谱峰,多次累加后蒸干,即得所述的两个蒽醌类化合物fistulaquinonea和fistulaquinoneb。
实施例2
取干燥的豆科苏木亚科决明属木本植物腊肠树(cassiafistula)的细枝5.0kg,粗粉碎至40目,用70%的丙酮超声提取4次,每次60分钟,提取液合并;提取液过滤,减压浓缩至体积的1/4;静置,滤除沉淀物,浓缩成130g的浸膏a;浸膏a用mci装柱,在浸膏a中加入320g的80%甲醇水溶解,用90%甲醇水3~8l洗脱,收集洗脱液,减压浓缩得到64.3g浸膏c,浸膏c中加130g丙酮,然后加入100目硅胶130g拌样,拌样后,用200目硅胶1200g装柱;用体积比分别为20:1,9:1,8:2,7:3,6:4和1:1的氯仿-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分,得到6个部分a-f,其中,对收集到的样品2部分13.2g,再重复硅胶柱层析,用体积比分别为9:1,8:2,7:3,6:4,1:1和1:2的乙醚-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分,得到6个部分b1-b6,其中第b3部分,即7:3部分约1.32g,再以50~80%的甲醇为流动相,流速12ml/min,21.2´250mm,5mm的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为254nm,每次进样55ml,分别收集13min及20min的色谱峰,多次累加后蒸干,即得所述的两个蒽醌类化合物fistulaquinonea和fistulaquinoneb。
实施例3
取实施例1或2制备的蒽醌类化合物fistulaquinonea,为红色胶状物;测定方法为:用核磁共振,结合其它波谱技术鉴定结构;
(1)紫外光谱(溶剂为甲醇),lmax(loge):210(4.14),250(3.75),280(3.67),408(3.26)nm;
(2)红外光谱(溴化钾压片)nmax:3348,3085,2947,1662,1625,1560,1447,1401,1365,1332,1280,1246,1198,1113,1062,1010cm-1;
(3)hresims显示本发明化合物准分子离子峰m/z349.1059[m+na]+(计算值为349.1052),结合13c和1hnmr谱(图1和图2,碳谱氢谱数据归属见图5)给出其分子式为c19h18o5,其不饱和度为11,1hnmr(c5d5n,500mhz)和13cnmr(c5d5n,125mhz)数据,见图5;
取实施例1或2制备的蒽醌类化合物fistulaquinoneb,为红色胶状物;测定方法为:用核磁共振,结合其它波谱技术鉴定结构;
(1)紫外光谱(溶剂为甲醇),lmax(loge):210(4.19),248(3.72),277(3.62),405(3.18)nm;
(2)红外光谱(溴化钾压片)nmax:3082,2945,1668,1620,1564,1443,1405,1358,1324,1283,1242,1196,1107,1054,996cm-1;
(3)hresims显示本发明化合物准分子离子峰m/z349.1046[m+na]+(计算值为349.1052),结合13c和1hnmr谱(图3和图4,碳谱氢谱数据归属见图5)给出其分子式为c19h18o5,其不饱和度为11,1hnmr(c5d5n,500mhz)和13cnmr(c5d5n,125mhz)数据,见图5。
实施例4
取实施例2制备的蒽醌类化合物fistulaquinonesa和b分别按实施例3中的方法进行结构测定,结果为:其结构同实施例3,分子式分别为c19h18o5和c19h18o5。
实施例5
取实施例1和2所制备的蒽醌类化合物进行抗烟草花叶病毒活性检测试验,试验情况如下:
采用半叶法,在药剂的质量浓度为50mg/l时对本发明化合物进行抗烟草花叶病毒性测定。在5~6龄烤烟的植株上,选取适用于测试的叶片(叶行正常,无病无虫),先将叶片均匀撒上细金刚砂,用毛笔将备用的烟草花叶病毒源(3.0×10-3)均匀抹在撒有细金刚砂的叶片上,待所有中选的叶片接毒结束后,立即放在盛有药液的培养皿中处理20min,取出,擦去叶片上水珠和药液,将两个半叶复原排放在铺有卫生纸保湿金的玻璃缸中,并盖上玻璃盖,控温(23±2)℃,放在室温自然光照射,2~3d即可见枯斑,每个处理都设另一半叶为对照,另外设有1组为商品南霉素的处理作为对比,按下列公式计算相对抑制率:
xi%=(ck-t)/ck×100%
x:相对抑制率(%),ck:浸泡于清水中半片接毒叶的枯斑数(个),t:浸泡于药液中半片接毒叶的枯斑数(个);
经对抗烟草花叶病毒的实验,fistulaquinonea和fistulaquinoneb的相对抑制率在20μm下分别达到25.2%和26.3%,低于阳性对照品南宁霉素的相对抑制率(28.8%),说明fistulaquinonea和fistulaquinoneb均具有一定的抗烟草花叶病毒活性。
实施例6
取实施例1和2所制备的蒽醌类化合物进行抗烟草花叶病毒活性检测试验,试验情况如下:
细胞株:白血病细胞(nb4)、肺癌细胞(a549)、人神经母细胞瘤细胞(shsy5y)、前列腺癌细胞(pc3)、乳腺癌细胞(mcf7)均由中国科学院上海药物研究所提供;
实验设计:以上细胞与不同浓度化合物温育72小时,每株细胞的实验均重复一次,用两次实验的结果进行数据处理,采用改良mtt法及srb法评价化合物对细胞增殖的抑制程度,计算抑制率,根据抑制率采用logit方法计算ic50,比较化合物的体外抗肿瘤活性;
细胞的增殖抑制率=(空白对照od值-加药孔的od值)/空白对照od值×100%;
(a)改良mtt法
取处于对数生长期的悬浮细胞,将细胞浓度调整为4×104/ml,加入96孔培养板,90µl/孔。阳性对照为顺铂,用生理盐水溶解;每孔分别加入10μl不同浓度的样品(1号试液-5号试液);加样组及对照组均设4个复孔,加样组、阳性对照组的高浓度组还设培养基的加药平行孔,每块板均设有4个空白对照孔(仅加培养基)样品的终浓度分别为10-2、10-1、1、10及102µg/ml,相应dmso的终浓度分别为0.1%、0.01%、0.001%、0.0001%、0.00001%;样品在终浓度102µg/ml时,用0.1%dmso作为溶剂对照,其余浓度均用生理盐水作阴性对照。阳性对照药顺铂的终浓度为10-1、1、10µg/ml;细胞在37℃,5%co2培养箱中分别孵育48h后,加入mtt(5mg/ml,sigma),10µl/孔;继续培养4h后,加入三联液[10%sds–5%异丁醇–0.012mol/lhcl(w/v/v)],100µl/孔,放置过夜后用酶标仪在570nm、630nm双波长下测定各孔的od值;
(b)srb法
取处于对数生长期的贴壁细胞株,用25%胰酶常规消化后,再用15%小牛血清完全rpmi-1640培养基将细胞浓度调整为5×104/ml,加入96孔培养板,90μl/孔。细胞在37℃,5%co2培养箱中分别孵育24h后加入阳性对照、阴性对照以及受试样品(各受试浓度同上mtt法,10μl/孔),样品的终浓度分别为10-2、10-1、1、10、102μg/ml,相应dmso的终浓度分别为0.1%、0.01%、0.001%、0.0001%、0.00001%;样品在终浓度102μg/ml时用0.1%dmso作为溶剂对照,其余浓度均用生理盐水作阴性对照;阳性对照药的终浓度为10-1、1、10μg/ml,阴性对照为等体积的生理盐水;加样组及对照组均设4复孔,加样组、阳性对照组的高浓度组还设培养基的加药平行孔,每块板均设有4个空白对照孔(仅加培养基);将96孔培养板置于37℃,5%co2培养箱中孵育(细胞与样品作用)48h后,加入4℃、50%的tca(三氯乙酸)50μl/孔;加完tca后,将96孔培养板置于4℃孵育1小时,取出培养板,轻轻倾去板内液体;用自来水轻轻冲洗5遍(将自来水由烧杯中轻轻倒入板中,轻晃后再将水倒去),置于空气中风干至不见水痕;然后加入配制好的0.4%srb(用1%乙酸稀释),50μl/孔,于室温下静置染色30分钟后倾去srb溶液,用1%乙酸冲洗4遍,以除去未与蛋白质结合的染料;置于空气中风干至无水痕后,加入10mm未缓冲tris(缓血氨酸)溶液150μl/孔(ph10,用三蒸水配制),将染料溶解后,于振荡器上振荡5分钟,用酶标仪在570nm波长下读取各孔od值;
(c)实验结果
实验结果表明:经对白血病急性早幼粒nb4细胞、肺腺癌a549细胞、人骨髓神经母细胞瘤shsy5y细胞、人前列腺癌pc3细胞、人乳腺癌mcf7细胞的细胞毒活性实验,蒽醌类化合物fistulaquinonea对nb4和shsy5y细胞株具有一定的细胞毒活性,ic50值分别达6.2和8.4μm;蒽醌类化合物fistulaquinoneb对a549细胞株具有一定的细胞毒活性,ic50值为5.5μm。
- 还没有人留言评论。精彩留言会获得点赞!