一种苯乙烯‑异戊二烯‑苯乙烯三嵌段共聚物均相催化加氢的方法与流程
(一)技术领域
本发明涉及热塑弹性体加氢技术领域,特别涉及一种苯乙烯-异戊二烯-苯乙烯三嵌段共聚物均相催化加氢的方法。
(二)
背景技术:
高分子不饱和聚合物由于含有碳碳不饱和双键,导致其容易氧化,耐候性和热稳定性不足,限制了其广泛的发展。其中最具代表性的是苯乙烯和丁二烯的嵌段共聚物(sbs),sbs通过加氢使双键饱和制得sebs,弥补了sebs的缺陷。但是遗憾的是sebs具有非极性,因而对极性的物质粘结性较差,尤其是对较硬和较粘稠的sebs进行软化和增粘是较困难的。据研究发现,sis与sbs相比具有诸多优异性能。首先,sis与其他材料的相容性优于sbs;其次sis具有特殊的流变性能,但是溶液粘度只有sbs的2/3;在抗老化性能上sis也优于sbs。同sbs一样,sis中橡胶相的不饱和双键在空气中会受到氧气、臭氧和紫外线的作用而发生热老化。进行选择性的加氢是解决这一问题的重要途径。sis选择加氢后成为seps,具有很多优异性能,尤其是在耐热性、抗老化和力学性能方面有很大的改进。
目前有关聚合物加氢的工艺技术,已有不少报道。例如,usp:4501857介绍了日本旭化成公司yasushikishimoto,ayase等较早用二氯双(环戊二烯基)钛作为催化剂对苯乙烯类弹性体sis氢化实现工业化生产。将sis溶于无水环己烷中配制成质量分数为5%的溶液,将1000g的高聚物溶液置于2l的干燥高压釜中,在氢气的氛围中保持40℃并不断地搅拌,再将20ml含有1.0mmol/100ml的二氯化双(环戊二烯基)钛的甲苯溶液加入,调节氢气压力为0.5mpa,反应2h后氢化度达到98%,苯环的加氢度小于1%,其中n(li)/n(ti)=4。如改变主催化剂的用量及li的含量,只要条件合适,它仍能使氢化度达到95%以上。可见二茂钛的化合物对嵌段共聚物的选择性加氢的效果很好;专利cn102580774a提供了一种不饱和聚合物加氢催化剂及其制备方法,由ia、iia、iiia族的金属有机化合物与镍/钴金属无机盐反应生成,有机金属/无机金属的金属摩尔比为1-10:1。该催化剂对共轭二烯的均聚物或共轭二烯与乙烯基芳烃的共聚物加氢效果显著,加氢选择性高,加氢反应后不易残留在聚合物中,可以水洗除去,有助于提高聚合物的纯度与品质;专利cn1191225a采用四丁基钛类化合物作为主催化剂(a),用带有一个或多个以上的酯基或羟基官能团的有机化合物作助催化剂(b)的高效催化体系进行选择加氢。每100g聚合物使用a0.05-0.2mmol,n(a):n(b)=1:(0.1-4.2),在一定的温度和压力下加氢反应0.5-1h,共轭二烯段加氢度达95%以上,该专利有效地解决了主催化剂在反应过程中活性下降问题,具有催化体系活性高、反应时间短等优点,适合于共轭二烯烃聚合物的选择加氢。
在上述各加氢过程都是介绍了同一类均相催化加氢的催化剂:(a)含有第ⅷ族元素的有机金属化合物或者是含有第ⅷ族元素的配合物;(b)结构式为ram的烷基金属化合物,r为c2-c10的取代/未取代的直链烷基或者是取代/未取代的支链烷基;m为金属阳离子,a为烷基基团的配位数与m的价态数相等。该类催化剂代替早期的多相催化剂,解决了多相催化剂加氢活性低的缺点,而且均相催化不易引起聚合物降解,凝胶量少;同时反应条件要求不高,无需高温高压以及无需大量催化剂,同时克服了多相催化选择性差的问题,该类催化剂选加氢择选择性强,一般烯烃段中双键加氢度可达98%以上,而乙烯基芳烃中苯环的加氢度小于2%。基于多相催化的众多缺点,逐步采用了均相催化。
(三)
技术实现要素:
本发明为了弥补现有技术的不足,提供了一种操作简单、无需脱除催化剂、加氢度高的苯乙烯-异戊二烯-苯乙烯三嵌段共聚物均相催化加氢的方法。
本发明是通过如下技术方案实现的:
一种苯乙烯-异戊二烯-苯乙烯三嵌段共聚物均相催化加氢的方法,以三嵌段共聚物sis为处理对象,其特征为,包括如下步骤:(1)将催化剂b溶液加入催化剂a溶液中,在40-100℃下陈化10-60min,得到加氢催化剂,其中,催化剂a为含有第ⅷ族元素的有机金属化合物和或含有第ⅷ族元素的配合物,催化剂b为结构式为ram的烷基金属化合物,ram中,r为取代/未取代的直链烷基或取代/未取代的支链烷基,a为配位数与m的价态数相等的烷基基团,m为金属阳离子;(2)将三嵌段共聚物sis溶于环己烷中,置于加氢反应釜,搅拌条件下抽真空后升温,向加氢反应釜中加入加氢催化剂,充入氢气进行1-10h的加氢反应;(3)反应结束后冷却至室温,取出加氢的胶液,并往胶液中滴入盐酸,再用无水乙醇絮凝,经真空干燥后得到产品。
本发明引用ni(naph)2/al(i-bu)3作为均相加氢催化剂,选择性对sis中的碳碳不饱和双键进行了加氢,并探索了最佳加氢条件。
所述三嵌段共聚物sis为质量浓度为1.5-3%的聚合物溶液,其溶剂为正戊烷、正辛烷、环己烷、正己烷、乙醚、庚烷、甲苯和苯中的一种或多种。
步骤(1)中,催化剂a和催化剂b分别用惰性溶剂溶解,得到催化剂a溶液和催化剂b溶液,催化剂a溶液的质量浓度为1-5%,催化剂b溶液的浓度为0.5-5mol/l;催化剂a和催化剂b的摩尔比为1:4-8;其中,所述惰性溶剂为烷烃、环烷烃和芳烃中的一种或多种,优选的是,惰性溶剂为正戊烷、正辛烷、环己烷、正己烷、乙醚、庚烷、甲苯和苯中的一种或多种。
加氢催化剂混合时,将催化剂b加入催化剂a溶液中,这样可以使al(i-bu)3将ni2+还原成有加氢活性的低价态的ni+和ni0。
催化剂a为有机酸的第ⅷ族金属盐,第ⅷ族元素选自铁、钴、镍和钯中的一种或者多种;有机酸为c2-c10的链烷酸或c3-c12的环烷酸;催化剂b的结构式ram中,r为c2-c6的取代/未取代的直链烷基或取代/未取代的支链烷基,a为乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、正戊基、正己基和环己基中的一种或多种,m为铝或锂。
其中,有机酸为c4-c8的链烷酸或c5-c10的环烷酸;优选的,链烷酸正丁酸、异丁酸、仲丁酸、叔丁酸、正戊酸、异戊酸、仲戊酸、叔戊酸、新戊酸、正己酸、仲己酸、正庚酸、仲庚酸、正辛酸、仲辛酸和2-乙基己酸中的一种或多种,环烷酸为环丙烷乙酸、环丁烷甲酸、环丁烷乙酸、环丁烷丙酸、环戊烷甲酸、环戊烷乙酸、环戊烷丙酸、2-甲基环戊烷甲酸、3-甲基环戊烷甲酸、3-甲基环戊烷乙酸、环己烷甲酸、环己烷乙酸和环庚烷甲酸中的一种或多种。
最优选的是,催化剂a为环烷酸镍,催化剂b为三异丁基铝、三乙基铝、正丁基锂和仲丁基锂中的一种或多种。
步骤(2)中,加氢反应釜中抽真空置换空气三次后升温至40-100℃,加氢催化剂的加入量为0.1-1mmol/g三嵌段共聚物sis,充入氢气至压力为1-5mpa,进行1-5h的加氢反应。
本发明无需专门脱除催化剂,实验操作简单易行,提高了催化剂效率,降低了催化剂用量,同时在较低温度下即可氢化高分子量的热塑性弹性体sis,并且还具有很好的加氢选择性,而且加氢度高达90%以上。
(四)附图说明
下面结合附图对本发明作进一步的说明。
图1为本发明实施例1产物的红外测试结果示意图;
图2为本发明实施例2产物的红外测试结果示意图;
图3为本发明实施例3产物的红外测试结果示意图;
图4为本发明实施例4产物的红外测试结果示意图;
图5为本发明实施例5产物的核磁谱图。
(五)具体实施方式
为了阐明本发明的技术的先进性和创新点,使本发明技术更加易于了解,结合具体事例作进一步的说明。
一、实验步骤:
1.首先是催化剂的配制
将3.7mol/l的环烷酸镍加入到干燥且为真空状态的聚合瓶中,然后向其中加入三异丁基铝溶液,二者发生氧化还原反应,环烷酸镍的溶液由绿色变为黑色。将ni(naph)2/al(i-bu)3催化剂放在40℃-80℃的水浴中进行陈化,陈化0.5h-1h后加入反应釜。
2.其次是无水无氧的加氢反应环境
将sis的环己烷溶液加入反应釜,抽真空换气至少三次。同时配制催化剂的聚合瓶也要抽真空通氮气至少三次,同时烤瓶保证聚合瓶干燥无水。待催化剂陈化结束后在氢气的压力下加入反应釜。整个反应过程中都能保证无水无氧。
二、下面通过实施例,对本发明的应用效果做具体说明。
实施例1:
配制了三种不同质量分数的sis胶液,变化范围在0.5%-3%,每种胶液质量分数下加入催化剂的相对含量不变,以镍的含量为标准设定0.1-1mmol/gsis,其中al/ni物质的量比为2-8,加氢压力设为1mpa-5mpa,加氢温度设为40℃-100℃,反应时间1h-5h。所得产物经红外测试,实验结果如附图1所示,各胶液质量分数下的加氢度如下表所示。
实施例2:
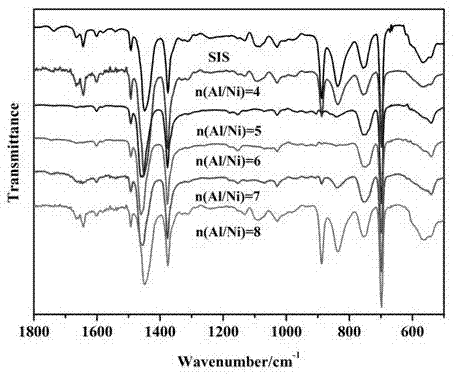
配制1.5w%的sis胶液,变化al/ni物质的量比范围在2-8,其他反应条件如实施例1,实验红外测试结果如附图2所示,所得加氢度如下表所示。
实施例3:
配制1.5w%的sis胶液,变化加氢温度为在40℃-100℃范围内,其他实验条件如实施例1,实验红外测试结果如附图3所示,各加氢温度所得加氢度如下表所示。
实施例4:
配制1.5w%的sis胶液,变化加氢压力在1mpa-5mpa范围内,其他实验条件如实施例1,实验测试结果如附图4所示,各加氢压力下所得加氢度如下表所示。
实施例5:
配制质量分数为0.5%-3%的sis胶液,加入催化剂的量以镍的含量为标准设定0.1-1mmolni/gsis,其中al/ni物质的量比为2-8,加氢压力设为1mpa-5mpa,加氢温度设为40℃-100℃,反应时间1h-5h。加氢反应后的sis做核磁测试,与加氢前的sis的核磁谱图比较如附图5所示,sis核磁谱图中1,4—加成,=ch—基团产生的化学位移在5.13ppm处,3,4—加成,=ch2基团产生的化学位移再5.13ppm和4.68ppm处,与之对比,氢化后的sis核磁谱图中5.13ppm、5.13ppm和4.68ppm处的谱峰变的很小甚至已经消失。
结果与讨论:
1.胶液的质量分数
变化sis的胶液质量分数为0.5%-3%,本实验所使用sis的数均分子量为18w,因此随着胶液质量分数的增大,分子链的流动性变差,对加氢效果影响显著,当胶液质量分数为3%时,基本未被加氢。
2.al/ni物质的量比
变化n(al)/n(ni)比为2-8,控制其他加氢反应条件不变。当n(al)/n(ni)比为4和8时加氢效果都相对较差,当n(al)/n(ni)比在5-7时加氢效果相对较好。实验结果证明:在n(al)/n(ni)比太小时,无法加氢或加氢效果不明显,随着比值的增大,加氢速度加快,加氢度提高,但太大的话又将导致加氢速度降低,加氢度下降。其原因为:al的量很少的话则不能将ni2+还原成有加氢活性的ni+和ni0,而ni2+本身对sis加氢反应没有催化作用。但当al的量过多,也几乎无催化作用,因为其改变了ni+和ni0比例关系使ni0含量增多甚至全为ni0,容易形成(ni0)n集体减少双键与活性中心配位几率,从而使催化活性下降。因此需要选择合适的比值。
3.加氢反应温度
变化加氢反应温度为40℃-100℃,控制其他加氢反应条件不变。温度越高,反应速度就越快。由于随着温度的升高,体系的黏度下降,从而增大了氢气的溶解度和扩散速度,同时也提高了反应活性中心的活性,加快了加氢反应的速度,但是如果温度过高,加氢度逐渐下降,这可能与分子在活性中心上活化吸附的几率随温度升高而降低有关也有可能是由于温度过高造成胶液的交联,甚者造成催化剂失活而影响加氢效果,因此当加氢反应温度达到80℃-100℃时加氢度降低。
4.加氢压力
变化加氢压力为1mpa-5mpa,控制其他加氢反应条件不变。在一定范围内变化加氢压力,整体来看对sis的加氢效果影响不大,但是随着加氢压力的增大,加氢度略微升高。同时为了验证在不同压力下的不同时间点的加氢度,做了三组实验,分别设定加氢压力为1mpa、3mpa和5mpa,每个压力下取了10个样,分别测定其加氢度,以1,4链节的加氢度的实验数据为例。实验结果显示反应初期压力对加氢度的影响显著,由于在反应初期随着氢气压力加大,氢气与催化剂形成的活性中心与双键接触越容易,所以反应初期,不同的加氢压力下所得的sis的加氢度不同。随着反应时间的延长,双键基本上都已与氢气与催化剂形成的活性中心接触,所以最终加氢度相差不大。
- 还没有人留言评论。精彩留言会获得点赞!