一种新型蓝光OLED材料及其制备方法与流程
本发明属于蓝光有机发光二极管材料领域,具体涉及一种新型蓝光oled材料,金刚烷改性聚3,6-咔唑的高分子化合物及其制备方法。
背景技术:
oled即有机发光二极管,又称为有机电激光显示、有机发光半导体。由美籍华裔教授邓青云于1979年在实验室中发现。oled显示技术具有自发光、广视角、几乎无穷高的对比度、较低耗电、极高反应速度等优点。
在oled的制备和优化中,发光材料的选择至关重要,其性质是决定了器件性能的重要因素之一。全色显示需要红绿蓝三基色的发光材料共同作用,其中蓝光材料由于稳定性、量子效率和色纯度等问题,制约了平板显示器的实用化进程。目前,聚咔唑及其衍生物是典型的蓝光材料,具有良好的光学和电学性质,被认为是最具应用潜力的高分子蓝光材料之一。然而大部分聚咔唑类发光材料的玻璃化转变温度tg较低,使器件的稳定性差,致使发光不稳定,器件寿命短,限制了咔唑材料的应用范围。
技术实现要素:
为克服现有蓝光oled材料稳定性的不足,本发明的目的在于提供了一种新型蓝光oled材料。该材料为金刚烷改性聚3,6-咔唑的高分子化合物,有较高的热稳定性,吸收光的带宽较宽,光学性能优异。
本发明的另一目的在于提供所述的新型蓝光oled材料的制备方法。该方法产率高,且易于操作和控制。
本发明提出一种新型蓝光oled材料,为金刚烷改性聚3,6-咔唑的高分子化合物,所述的金刚烷改性聚3,6-咔唑的高分子化合物具有如下的结构通式:
其中,p、q为单元组分的摩尔分数,满足:p+q=1,0.5≤p<1.0,r为氢或具有1-22个碳原子是烷烃。
优选的,所述的p=0.9,q=0.1或p=0.7,q=0.3或p=0.5,q=0.5。
所述的金刚烷改性聚3,6-咔唑的高分子化合物的数均分子量为3500-6000,分子量分布为1.05-1.3。
一种上述的新型蓝光oled材料的制备方法,包括如下步骤:将n-烷基-3,6-二硼酸酯咔唑单体、n-烷基-3,6-二溴咔唑和1,3-二(4-溴苯基)金刚烷按摩尔比1:0~0.5:0~0.5溶于溶剂中,然后加入弱碱水溶液,通入氮气,加入催化剂四(三苯基膦)钯,油浴加热至30-150℃,反应1-7天后,得到反应混合物;将所述的反应混合物滴入无水甲醇中,过滤,丙酮索式提取,真空干燥,得到金刚烷改性聚3,6-咔唑的高分子化合物;所述的n-烷基-3,6-二硼酸酯咔唑单体和弱碱的摩尔比为1:1-10,所述的催化剂的用量为所有反应物总摩尔数的0.05-10%。
所述n-烷基-3,6-二溴咔唑的制备方法,包括如下步骤:
s1、3,6-二溴咔唑的合成;
在冰水浴中,向反应器皿中加入咔唑和n,n-二甲基甲酰胺溶剂;n-溴代丁二酰亚胺和n,n-二甲基甲酰胺的混合溶液经恒压滴液漏斗逐滴加入到所述反应器皿中;滴完完毕后,将反应器皿中的反应混合物升温至100°反应0.5h;反应结束后,将反应混合物倒入冰水中,搅拌抽滤,收集析出的浅绿色固体,洗涤,真空干燥,经无水乙醇重结晶得固体3,6-二溴咔唑,所述咔唑与n-溴代丁二酰亚胺的摩尔比为1:2~3;
s2、n-烷基-3,6-二溴咔唑的合成;
冰浴条件下,将步骤s1制得的3,6-二溴咔唑溶于无水溶剂中,并缓慢分批加入氢化钠;缓慢升温至25℃室温,搅拌60min后,经恒压滴液漏斗滴加溴代烷烃和n,n-二甲基甲酰胺的混合溶液;所述3,6-二溴咔唑与所述溴代烷烃的摩尔比为1:1.1~1.5;室温搅拌反应24h后,加入冰水猝灭结束反应;经萃取,干燥,减压浓缩蒸馏得固体产品粗品;经柱层析硅胶柱分离提纯得白色固体n-烷基-3,6-二溴咔唑,其结构通式为:
所述n-烷基-3,6-二硼酸酯咔唑单体的制备方法,包括如下步骤:
s3、在无水无氧条件下,加入所述n-烷基-3,6-二溴咔唑和无水溶剂,氮气脱气处理三次后,置于-78℃低温反应浴中,搅拌30min;缓慢加入正丁基锂,滴加完毕后,反应混合物继续搅拌2h;加入异丙醇频哪醇硼酸酯,搅拌60min后,缓慢升温至25℃室温,继续反应24h;所述n-烷基-3,6-二溴咔唑、所述正丁基锂与所述异丙醇频哪醇硼酸酯的摩尔比为1:2~5:2~5;反应结束后,冰水猝灭,并用无水乙醚萃取有机相;有机相经氯化钠溶液洗涤后,无水硫酸镁干燥,减压蒸馏浓缩,真空干燥可得固体产品粗品;粗产品经硅胶层析柱纯化得白色固体n-烷基-3,6-二硼酸酯咔唑单体,其结构通式为:
所述1,3-二(4-溴苯基)金刚烷的制备方法,包括如下步骤:
s4、1,3-二溴金刚烷的合成;
将金刚烷和液溴的混合物冷却到0℃,加入润湿的铁粉室温反应3h后,倒入冰水中,过滤,取滤渣,经饱和亚硫酸氢钠溶液和蒸馏水洗涤,干燥,乙醇重结晶,得到1,3-二溴金刚烷;
s5、1,3-二(4-溴苯基)金刚烷的合成;
将步骤s4的1,3-二溴金刚烷和无水三氯化铁、溴苯依次加入反应器皿中,回流加热至165℃,反应结束后,二氯甲烷萃取有机相,洗涤,干燥,过滤,浓缩,硅胶柱层析提纯,得1,3-二(4-溴苯基)金刚烷;所述的1,3-二溴金刚烷与溴苯的摩尔比为1:10-30。
所述的溶剂为四氢呋喃或甲苯中的一种或多种。
所述的弱碱水溶液为碳酸钠、碳酸钾或碳酸氢钠的水溶液。
本发明的有益效果:本发明通过在聚3,6-咔唑中引入金刚烷,使热稳定性,和光学性能都得到提升。具体为提高聚合物的吸光发光效果,减少蓝色荧光猝灭的发生,所以改善了蓝光oled材料的荧光性能和光电效率。且本发明的高分子化合物的成膜性能较好,溶解性能优良。本发明还提供所述的新型蓝光oled材料的制备方法,易于操作和控制,产率≥90%。
附图说明
图1为本发明的3,6-二溴咔唑的红外光谱图。
图2为本发明的3,6-二溴咔唑的核磁共振氢谱图。
图3为本发明的3,6-二溴-9-(2-乙基己基)咔唑的红外光谱图。
图4为本发明的3,6-二溴-9-(2-乙基己基)咔唑的核磁共振氢谱图。
图5为本发明的n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑的红外光谱图。
图6为本发明的n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑的核磁共振氢谱图。
图7为本发明的1,3-二溴金刚烷的红外光谱图。
图8为本发明的1,3-二(4-溴苯基)金刚烷的红外光谱图。
图9为本发明的1,3-二(4-溴苯基)金刚烷的核磁共振氢谱图。
图10为本发明的实施例一至三的产物p1、p2和p3的红外光谱图。
图11为本发明的实施例一至三的产物p1、p2和p3的核磁共振氢谱图。
具体实施方式
如下结合附图,对本申请方案作进一步描述:
实施例一
1、n-烷基-3,6-二溴咔唑和n-烷基-3,6-二硼酸酯咔唑单体的制备
1.1、3,6-二溴咔唑的合成,反应式如下:
在冰水浴中,向150ml的三口圆底烧瓶中加入咔唑(3.35g,20mmol)和40mln,n-二甲基甲酰胺溶剂。n-溴代丁二酰亚胺(7.59g,43mmol)和16mln,n-二甲基甲酰胺的混合溶液经恒压滴液漏斗逐滴加入到所述烧瓶中。滴完完毕后,将烧瓶中的反应混合物升温至100°反应0.5h。反应结束后,将反应混合物倒入300ml冰水中,搅拌抽滤,收集析出的浅绿色固体,并用去离子水洗涤多次,真空干燥。粗品经无水乙醇重结晶得固体3,6-二溴咔唑4.77g,产率为73.4%。
1.2、3,6-二溴-9-(2-乙基己基)咔唑的合成,反应式如下:
在置于冰浴条件下的100ml三口圆底烧瓶中,将3,6-二溴咔唑(2g,6.15mmol)溶于无水处理的四氢呋喃(20ml)溶剂中,并缓慢分批加入氢化钠(346mg,60%w/w分散于矿物油中,8.6mmol)。缓慢升温至室温(25℃),搅拌60min后,经恒压滴液漏斗滴加2-乙基己基溴(1.43ml,8mmol)和n,n-二甲基甲酰胺(4ml)的混合溶液。室温搅拌反应24h后,加入冰水猝灭结束反应。反应混合物依次用二氯甲烷萃取,无水硫酸镁干燥,减压浓缩蒸馏得固体产品粗品。经柱层析硅胶柱分离提纯(流动相v正己烷:v乙酸乙酯=10:1,rf=0.8),得白色固体3,6-二溴-9-(2-乙基己基)咔唑2.41g,产率为89.4%。
1.3、n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑的合成,反应式如下:
在100ml无水无氧反应瓶中,加入3,6-二溴-9-(2-乙基己基)咔唑(1g,2.29mmol)和无水四氢呋喃(18ml),氮气脱气处理三次后,置于低温反应浴-78℃,搅拌30min。经注射器缓慢加入正丁基锂(1.8ml,2.7m),滴加完毕后,反应混合物继续搅拌2h。异丙醇频哪醇硼酸酯(1.05g,5.4mmol)经注射器迅速加入无水无氧反应瓶中,搅拌60min后,缓慢升温至室温,继续反应24h。反应结束后,冰水猝灭,并用无水乙醚萃取有机相。有机相经氯化钠溶液洗涤后,无水硫酸镁干燥,减压蒸馏浓缩,真空干燥可得固体产品粗品。粗产品经硅胶层析柱纯化(流动相v正己烷:v乙酸乙酯=25:1,rf=0.25),得白色固体n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑0.73g,产率60.7%。
2、1,3-二(4-溴苯基)金刚烷单体的制备
2.1、1,3-二溴金刚烷的合成,反应式如下:
向500ml三口圆底烧瓶中依次加入金刚烷(27.2g,0.2mol)和液溴(50ml)。在冰水溶液中使反应混合物冷却到0℃后,加入铁粉(0.75g,13.4mmol,已用几滴水润湿),此时反应开始,生成红棕色气体。反应3h后,将所得的反应物倒入冰水溶液中,过滤,再经经饱和亚硫酸氢钠溶液、蒸馏水洗涤,干燥后得固体产品粗品。继续用95%的乙醇重结晶得无色晶体39.4g,产率67.1%。
2.2、1,3-二(4-溴苯基)金刚烷的合成,反应式如下:
向100ml三口烧瓶中依次加入1,3-二溴金刚烷(2.94g,10.0mmol)、无水三氯化铁(64.8mg,0.4mmol)和重蒸溴苯(25.12g,160mmol)。油浴缓慢升温到165℃,加热回流16h。待结束反应,将反应得到的混合物加入50ml二氯甲烷溶液和50ml0.1mol/l的hcl混合溶液中。用饱和碳酸氢钠溶液洗涤有机层,继续用无水硫酸镁干燥,过滤,浓缩。硅胶过柱柱层析提纯(正己烷,rf=0.4),得无色晶体2.28g,产率51.2%。
3、金刚烷改性聚3,6-咔唑的高分子化合物p1的制备
在100ml无水无氧反应瓶中,依次加入3,6-二溴-9-(2-乙基己基)咔唑(139.9mg,0.32mmol)、n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑(212.5mg,0.4mmol)和1,3-二(4-溴苯基)金刚烷(35.7mg,0.08mmol),甲苯(15ml)、甲基三辛基氯化铵(2滴)和碳酸钾水溶液(2mol/l,5ml)依次加入反应瓶后,无氧脱气处理三次。氮气保护下,催化剂四(三苯基膦)钯(12mg)加入至反应底物中。油浴加热升温至反应温度90℃,反应72h。反应结束后,将反应混合物滴入无水甲醇中,过滤,所得固体按无水甲醇、蒸馏水、无水甲醇的顺序洗涤多次。真空干燥后,采用丙酮索氏提取移除未反应的单体、低聚物和催化剂(72h)。真空干燥72h后得金刚烷改性聚3,6-咔唑的高分子化合物棕黄色粉末,产率≥90%。
实施例二
1、n-烷基-3,6-二溴咔唑和n-烷基-3,6-二硼酸酯咔唑单体的制备
1.1、3,6-二溴咔唑的合成,与实施例一相同。
1.2、3,6-二溴-9-(2-乙基己基)咔唑的合成,与实施例一相同。
1.3、n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑的制备,与实施例一相同。
2、1,3-二(4-溴苯基)金刚烷单体的制备
2.1、1,3-二溴金刚烷的合成,与实施例一相同。
2.2、1,3-二(4-溴苯基)金刚烷的合成,与实施例一相同。
3、金刚烷改性聚3,6-咔唑的高分子化合物p2的制备
在100ml无水无氧反应瓶中,依次加入3,6-二溴-9-(2-乙基己基)咔唑(46.6mg,0.16mmol)、n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑(212.5mg,0.4mmol)和1,3-二(4-溴苯基)金刚烷(107.1mg,0.24mmol),甲苯(15ml)、甲基三辛基氯化铵(2滴)和碳酸钾水溶液(2mol/l,5ml)依次加入反应瓶后,无氧脱气处理三次。氮气保护下,催化剂四(三苯基膦)钯(12mg)加入至反应底物中。油浴加热升温至反应温度90℃,反应72h。反应结束后,将反应混合物滴入无水甲醇中,过滤,所得固体按无水甲醇、蒸馏水、无水甲醇的顺序洗涤多次。真空干燥后,采用丙酮索氏提取移除未反应的单体、低聚物和催化剂(72h)。真空干燥72h后得金刚烷改性聚3,6-咔唑的高分子化合物棕黄色粉末,产率≥90%。
实施例三
1、n-烷基-3,6-二溴咔唑和n-烷基-3,6-二硼酸酯咔唑单体的制备
1.1、3,6-二溴咔唑的制备,与实施例一相同。
1.2、3,6-二溴-9-(2-乙基己基)咔唑的制备,与实施例一相同。
1.3、n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑的制备,与实施例一相同。
2、1,3-二(4-溴苯基)金刚烷单体的制备
2.1、1,3-二溴金刚烷的制备,与实施例一相同。
2.2、1,3-二(4-溴苯基)金刚烷的制备,与实施例一相同。
3、金刚烷改性聚3,6-咔唑的高分子化合物p3的制备
在100ml无水无氧反应瓶中,依次加入n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑(212.5mg,0.4mmol)和1,3-二(4-溴苯基)金刚烷(178.5mg,0.4mmol),甲苯(15ml)、甲基三辛基氯化铵(2滴)和碳酸钾水溶液(2mol/l,5ml)依次加入反应瓶后,无氧脱气处理三次。氮气保护下,催化剂四(三苯基膦)钯(12mg)加入至反应底物中。油浴加热升温至反应温度90℃,反应72h。反应结束后,将反应混合物滴入无水甲醇中,过滤,所得固体按无水甲醇、蒸馏水、无水甲醇的顺序洗涤多次。真空干燥后,采用丙酮索氏提取移除未反应的单体、低聚物和催化剂(72h)。真空干燥72h后得金刚烷改性聚3,6-咔唑的高分子化合物棕黄色粉末,产率≥90%。
一、上述实施例的产物的结果主要通过核磁共振氢谱(1hnmr)、红外光谱(ft-ir)和元素分析进行表征,具体如下:
(1)3,6-二溴咔唑
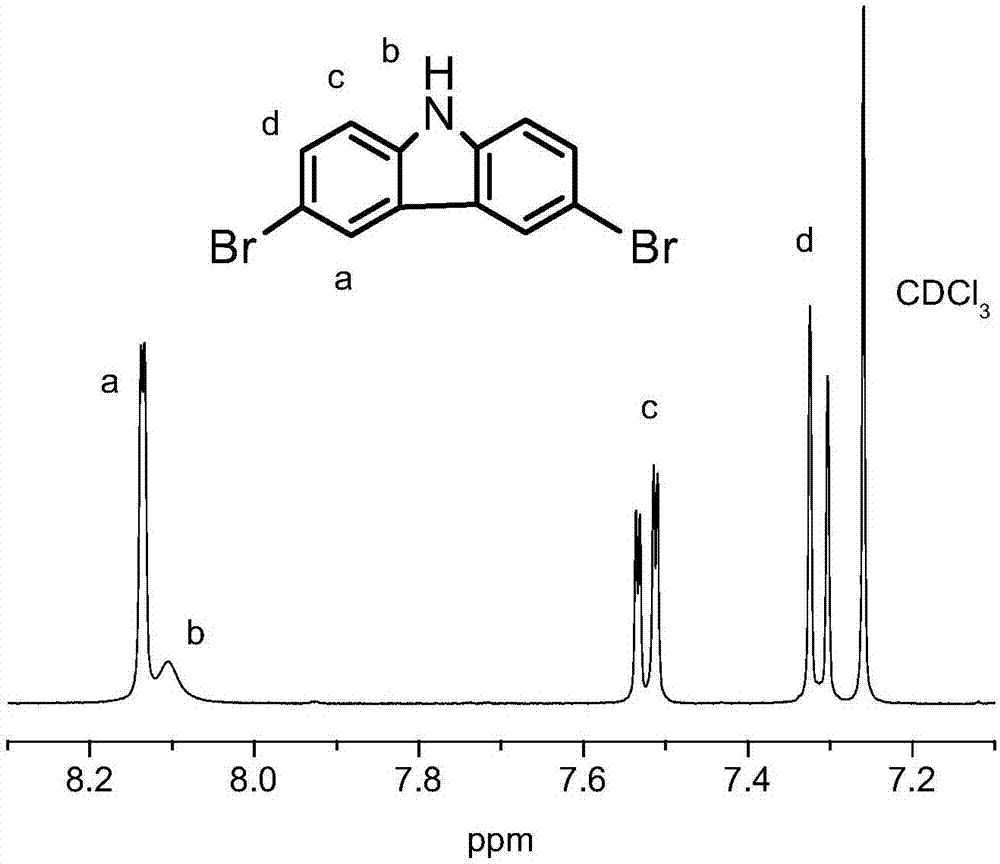
1hnmr(400mhz,dmso-d6):8.14(d,j=1.9hz,2h),8.10(s,1h),7.51(dd,j=8.6,1.9hz,2h),7.31(d,j=8.6hz,2h).ft-ir(kbr,cm-1):3420,1472,1432,1286,1240,1126,1052,899,867,803,565.anal.calcdforc12h7nbr2:c,44.35;h,2.17;n,4.31;br,49.17.found:c,44.42;h,2.30;n,4.25;br,49.03。
参见图1,图中明显可以看到n-h伸缩振动强吸收振动峰出现在3420cm-1,芳基碳(c-n)伸缩振动峰则出现在1286cm-1和1240cm-1,说明产物a中存在咔唑结构。565cm-1处的强吸收峰可以归属为c-br键的伸缩振动,产物a可以初步确定为3,6-二溴咔唑。在核磁共振氢谱,图2中,a,b,c,d四组峰的积分面积比例为2:1:2:2,质子总数目与比例与产物结构式相符。氢原子b单峰,化学位移值8.10,归属为与n直接相连的氢原子(n-h结构)。a,c,d处氢原子均为双峰,符合峰的裂分定律。结合红外光谱图和核磁共振氢谱图可得产物3,6-二溴咔唑顺利合成。元素分析结果符合理论计算值。
(2)3,6-二溴-9-(2-乙基己基)咔唑
3,6-二溴-9-(2-乙基己基)咔唑(b):1hnmr(400mhz,chloroform-d):8.13(d,j=1.9hz,2h),7.54(dd,j=8.7,1.9hz,2h),7.25(d,j=8.9hz,2h),4.10(dd,j=7.6,1.9hz,2h),1.99(hept,j=6.3hz,1h),1.29(m,8h),0.87(dt,j=17.6,7.2hz,6h).ft-ir(kbr,cm-1):2957,2927,2857,1471,1437,1378,1344,1317,1287,1218,1204,1148,1059,1018,866,832,796,644,564.anal.calcdforc20h23nbr2:c,54.94;h,5.30;n,3.20;br,36.56.found:c,54.84;h,5.50;n,3.33;br,36.33。
参见图3可知,烷烃碳氢(c-h)伸缩振动吸收峰出现于2957cm-1,2927cm-1和2857cm-1附近,且均为强峰。咔唑环仲胺基(n-h)吸收峰消失不见,说明烷基长链2-乙基己基成功将3,6-二溴咔唑的n原子取代。在核磁共振氢谱,图4中,各峰积分面积比例从左至右为2:2:2:2:1:8:6,质子总数目、比例、峰型均符合结构式。各峰的归属如图4所示。3,6-二溴咔唑(n-h)单峰从8.10处消失,在4.10-0.87处出现烷基2-乙基己基(-c8h17)的吸收峰,也说明3,6-二溴-9-(2-乙基己基)-9h-咔唑的顺利合成,与红外谱图一致。元素分析结果符合产物b的理论计算值。
(3)n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑
n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑(c):1hnmr(400mhz,chloroform-d):8.59(s,2h),7.82(dd,j=8.2,1.2hz,2h),7.30(d,j=8.3hz,2h),4.10(dd,j=7.5,3.0hz,2h),1.98(t,j=12.2,6.2hz,1h),1.24(m,32h),0.92(m,6h).ft-ir(kbr,cm-1):2976,2931,2874,1630,1597,1470,1380,1352,1319,1270,1256,1215,1146,1084,964,859,808,674.anal.calcdforc32h47o4nb2:c,72.33;h,8.91;n,2.64.found:c,72.42;h,9.13;n,2.71。
参见图5,碳溴键(c-br)吸收峰564cm-1消失,1352和1380cm-1处明显出现吸收强峰,这可以归结为b-o键的引入,4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基将溴原子成功取代。此外,受电负性影响,不受溴原子取代的苯环碳碳双键振动吸收峰移向1600cm-1附近,其余剩下各峰振动吸收波长,与产物b类似,可以认为产物c顺利合成。如图6所示,在核磁共振氢谱中,各组分积分面积比例从左至右依次为2:2:2:2:1:32:6,质子氢总数、比例及各峰峰型均符合产物d结构式。此外,对比产物b的核磁共振氢谱也可知,化学位移为1.39处的积分比例增大,其余不变,咔唑苯环两侧溴被取代,成功合成了3,6-咔唑硼酸化产物。元素分析结果符合产物n-(2-乙基己基)-3,6-二(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)咔唑的理论计算值。
(4)1,3-二溴金刚烷
1hnmr(400mhz,chloroform-d):2.87(s,2h),2.38–2.19(m,10h),1.70(t,j=2.9hz,2h).anal.calcdforc10h14br2:c,40.85;h,4.80;br,54.35.found:c,40.99;h,5.01;br,54.00。
如图7所示,a,b,c三处的峰面积比例为2:10:2,与1,3-二取代金刚烷的质子氢数目和比例相符。a处氢原子受相邻碳被溴原子取代的影响,低场移动,在2.87ppm处出现单峰。c处氢原子受溴原子诱导效应的影响最弱,在1.70ppm处出现多重峰。其余伯碳、仲碳氢原子(b)在2.19~2.38ppm处出峰。元素分析结果符合产物d的理论计算值。综上,可以认为产物1,3-二溴金刚烷成功合成。
(5)1,3-二(4-溴苯基)金刚烷
1hnmr(400mhz,chloroform-d):7.49–7.39(d,j=8.3hz,4h),7.32–7.18(d,j=8.3hz,4h),2.31(m,2h),1.95(s,2h),1.91(d,j=3.1hz,8h),1.77(t,j=3.1hz,2h).ft-ir(kbr,cm-1):3079,3050,2915,2901,2848,1491,1446,1394,1343,1075,1006,805,792,748,715,541,527.anal.calcdforc22h22br2:c,59.22;h,4.97;br,35.81.found:c,59.28;h,5.04;br,35.68。
1,3-二(4-溴苯基)金刚烷的红外光谱图如图8所示。由图可知,芳环骨架碳氢伸缩振动峰出现于3080cm-1附近,芳环碳碳骨架伸缩振动吸收峰约为1490cm-1。此外,烷基伸缩振动吸收峰出现于2915cm-1和2848cm-1附近,说明了1,3-二(4-溴苯基)金刚烷中存在了苯环结构和金刚烷结构。1,4-取代的苯环变形振动吸收峰出现于805cm-1附近,碳溴键(c-br)的伸缩振动吸收峰出现于527cm-1附近,说明了4-溴苯与金刚烷发生了1,3-二取代反应。在核磁共振氢谱图9中,金刚烷在1.95ppm处出现单峰,积分氢个数为2,同样证明了金刚烷发生了二取代反应。苯环在7.2-7.4ppm附近出现两个双峰,积分比例为1:1,证明苯环取代反应发生在对位。元素分析结果符合产物e的理论计算值。综上,可以认为产物1,3-二(4-溴苯基)金刚烷成功合成。
(6)金刚烷改性聚3,6-咔唑的高分子化合物p1、p2和p3
如图10所示,从对比图中可以观察到,p1、p2和p3彼此具有相似的特征吸收峰。p1的特征吸收峰:3053,2956,2927,2870,1630,1604,1471,1379,1344,1322,1268,1217,1203,1149,1073,874,796cm-1。p3的特征吸收峰:3057,2955,2923,2848,1631,1603,1480,1384,1349,1325,1286,1219,1153,1006,802cm-1。位于2800~2960cm-1范围的特征吸收峰可以归属为咔唑n-烷基长链和金刚烷烷基的碳氢伸缩振动吸收峰;同时,p1至p3位于2848cm-1与2878cm-1吸收峰的强度比例逐步提高,暗示了聚合物中金刚烷含量的提高。芳环碳碳骨架伸缩振动峰出现在1630cm-1和1600cm-1附近,也随着苯基金刚烷含量的提高而提高。芳香胺c-n伸缩振动吸收峰处于1322cm-1附近,表明聚合物中咔唑结构的存在。芳烃c-h变形振动,对位二取代特征峰(800cm-1附近)强度逐步增强,间位二取代特征峰(875cm-1附近)强度逐渐降低,进一步说明了金刚烷单体与咔唑单体成功聚合。综上所述,聚合反应顺利进行,共聚物p1~p3金刚烷含量如预期增加。
p1、p2和p3的结构与比例组成可由核磁共振氢谱图(1hnmr)进行进一步确认。对比p1、p2和p3及其单体核磁共振氢谱图,p3可以简单归属为:1hnmr(400mhz,chloroform-d):8.34(d,2h),7.81-7.60(m,4h),7.60-7.34(m,5h),7.32-7.25(m,2h),4.19(m,2h),2.36(m,2h),2.28–1.72(m,11h),1.49–1.10(m,8h),1.05–0.71(m,6h),如图11所示。咔唑环质子氢受n原子诱导效应的影响,化学位移较苯环质子氢稍高,出现于7.60ppm的右侧;同时随着金刚烷含量的增加,咔唑环质子氢化学位移整体向高场移动。金刚烷叔碳氢质子峰出现于2.36ppm附近,咔唑长链烷基的末端甲基质子峰(k)出现于1.05-0.71ppm附近。由此明显特征峰,可推断出共聚物中金刚烷的摩尔含量为p1(9.4mol%)、p2(28.1mol%)和p3(46.4mol%)。在3,6-咔唑共聚物中,金刚烷的实际摩尔含量均低于聚合物反应投料比,说明3,6-二溴-9-(2-乙基己基)-9h-咔唑的反应活性稍高于1,3-二(4-溴苯基)金刚烷单体。这很可能与金刚烷大体积因素和反应催化剂四(三苯基膦)钯对反应底物的选择性有关。
二、金刚烷改性聚3,6-咔唑的高分子化合物p1、p2和p3分子量的测定
表1不同投料比聚合物凝胶色谱仪测试结果
注:(1)投料含量指金刚烷在反应物中所占的摩尔含量;(2)实际含量指通过核磁共振氢谱确定的金刚烷在聚合产物中所占的摩尔含量。
实施例一至三的金刚烷改性聚3,6-咔唑的高分子化合物p1、p2和p3的凝胶色谱仪测试结果如表1所示。由表1可知,共聚物的数均分子量约为3500-6000左右,分散率pdi约为1.2附近。聚合产物数均分子量随着金刚烷摩尔含量的增加而逐渐升高,聚合产物的分散率则随之变窄,聚合产物p3降低尤为明显。金刚烷单体插入3,6-咔唑聚合物主链后,咔唑烷基长链间的排斥力减小,聚合反应能够平稳有序的进行,数均分子量增大,pdi分布变窄。聚合物p3,n-烷基-3,6-二硼酸酯咔唑单体与1,3-二(4-溴苯基)金刚烷交替连接,相比于p1和p2的无规共聚,其空间位阻降低最明显,pdi分布突然明显降低。p1和p2分散率相差较小,原因可能在于反应聚合单体活性的影响。1,3-二(4-溴苯基)金刚烷可能在聚合物主链上连续偶联,对聚合物的有序程度影响不明显。
上述优选实施方式应视为本申请方案实施方式的举例说明,凡与本申请方案雷同、近似或以此为基础作出的技术推演、替换、改进等,均应视为本专利的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!