一种单细胞甲基化测序文库的构建方法及其应用与流程
本发明涉及基因检测领域。具体地说,涉及一种单细胞全基因组甲基化测序文库的构建方法及其应用。
背景技术:
dna甲基化是基因组dna的一种重要表观遗传方式,是在dna甲基转移酶(dnamethyltransferase,dnmt)的催化下,以s-腺苷甲硫氨酸(s-adenosylmethionine,sam)为甲基供体,将甲基转移到特定的碱基上的过程。dna甲基化现象广泛存在于细菌、植物和动物中,参与生物体的多种生物学过程(lawjaandjacobsense,establishing,maintainingandmodifyingdnamethylationpatternsinplantsandanimals[j].natrevgenet,2010.11(3):204-220.)。在植物中,dna甲基化具有物种、组织、器官、细胞、发育阶段等时空特异性,在植物生长发育、适应环境条件以及进化过程中起着重要的调节作用(fujimotor,sasakit,kudohh,etal.,epigeneticvariationinthefwagenewithinthegenusarabidopsis[j].plantj,2011.66(5):831-843.)。
表观遗传学的研究现已成为热点,用于检测dna甲基化的方法也随之不断涌现。常用检测方法包括基于凝胶分析方法、基于基因芯片分析、基于高通量测序方法、高效液相色谱柱(highperformanceliquidchromatography,hplc)、甲基化敏感性限制性内切酶(ms-re)-pcr/southern法、结合亚硫酸氢盐的限制性内切酶法(combinedbisulfiterestrictionanalysis,cobra)、甲基化荧光检测(methylight)、限制性标记基因组扫描(restrictionlandmarkgenomicscanning,rlgs)和mbd柱层析与ms-re联用技术(combinationofmethylated-dnaprecipitationandmethylation-sensitiverestrictionenzymes,compare-ms)等。目前已报道的单细胞dna甲基化测序的方法有两种:一种是单细胞简化代表性重亚硫酸盐测序(single-cellreduced-representationbisulfitesequencing,scrrbs)(guoh,zhup,wux,etal.,single-cellmethylomelandscapesofmouseembryonicstemcellsandearlyembryosanalyzedusingreducedrepresentationbisulfitesequencing[j].genomeres,2013.23(12):2126-2135.)和单细胞重亚硫酸盐测序(single-cellbisulfitesequencing,scbs)(smallwoodsa,leehj,angermuellerc,etal.,single-cellgenome-widebisulfitesequencingforassessingepigeneticheterogeneity[j].natmethods,2014.11(8):817-820.)。scrrbs是第一个发明出来的单细胞甲基化测序方法。该方法采用先酶切cg位点再重亚硫酸盐转换并富集扩增的策略,仅能低覆盖率地检测gc酶切位点附近的甲基化;植物中由于存在大量的chg和chh甲基化类型,所以scrrbs并不适合植物中的分析。scbs操作原理是先对基因组进行重亚硫酸盐转换、片段化,然后再用随机引物对小片段进行两步扩增,能获得相比scrrbs更大的覆盖度及更无偏的甲基化位点信息,但亚硫酸盐转换产生的小片段的长度直接影响了文库的覆盖度(过长和过短的片段都无法进行测序)。
较之前的scrrbs和scbs技术,本发明中涉及的基于多重置换扩增的单细胞重亚硫酸盐测序(multipledisplacementamplificationsingle-cellbasedbisulfitesequencing,mb-scbs,简称mb-seq),可以忽略重亚硫酸盐转换产生的片段长短对文库片段长度的影响,利用该方法构建的测序文库的丰度和比对率更高,可适用于动物、微生物,尤其是植物的单细胞甲基化测序。
技术实现要素:
本发明的主要目的在于提供一种单细胞全基因组dna甲基化测序文库的构建方法,以及用于生产单细胞基因组dna甲基化测序的试剂盒,以提高单细胞尤其是来源于植物的单细胞全基因组dna甲基化测序文库的覆盖度和比对率,同时提高测试数据的重复性和稳定性。
为了实现本发明目的,本发明先利用优选的重亚硫酸盐转换试剂盒产生含尿嘧啶的小片段,后用无标签的随机引物进行数轮扩增,将游离的单链小片段引物洗去后,利用高温将配对双链变性,再将单链小片段连接成单链长片段,利用5’末端封闭的无标签随机引物片段进行mda扩增,获得扩增终产物,最终对反应产物进行基于tn5酶的文库构建。
具体来说,本发明方法包括如下步骤:
1)单细胞转化dna的获得;
2)dna的富集;
3)富集dna的建库;
4)文库定量和上机测序。
其中步骤1)进一步包括:①分离单个细胞或细胞核;②将单细胞或细胞核进行裂解,得到单细胞样本;③将未甲基化的胞嘧啶转化成尿嘧啶;④纯化产物dna片段。
其中步骤2)进一步包括:⑤短片段富集合成反应;⑥磁珠纯化并去除单链引物;⑦酶连反应;⑧多重置换扩增(mda);⑨磁珠纯化。
其中步骤3)进一步包括:⑩扩增产物浓度测定;
该发明方法中步骤1)、2)和3)的组合是必要的:单细胞的转化dna获得(步骤1)后,使用本方法的dna富集步骤是非常有效的(步骤2),富集后的产物只能搭配tn5基于的建库步骤建库测序(步骤3)。其中步骤1)-2)将mda的扩增方法引入了重亚硫酸盐测序的检测方法。步骤3)是基于tn5的dna建库试剂盒(trueprepdnalibraryprepkitv2for
其中步骤①中以27%的山梨醇溶液平衡胞内渗透压,维持正常的细胞形态。利用显微操作系统,单个细胞可以被分离到,能够用于dna重亚硫酸盐测序的检测。
步骤2)先将小片段富集,当浓度累积到一定程度以后,除去单链的随机引物小片段,将小片段酶连成长片段以后再进行mda扩增。这样的操作利用重亚硫酸盐转化后的小片段富集。
步骤⑤用不含标签的随机序列作为引物的。由于引物上不带有含barcode的特异序列,降低了运算量和无谓的成本,提高了数据的准确性。
步骤⑧利用不含标签的5’末端生物素修饰的随机序列作为引物,由于生物素修饰对5’末端的封闭作用,避免了t4rnaligase将随机引物链接成长链,可以减少非特异性链的产生。
附图说明
图1.mb-seq的操作流程图。灰色表示原始的dna单链,黑线表示加入的引物和利用引物合成的dna片段,小圆点表示引物及序列末端的生物素修饰封闭。
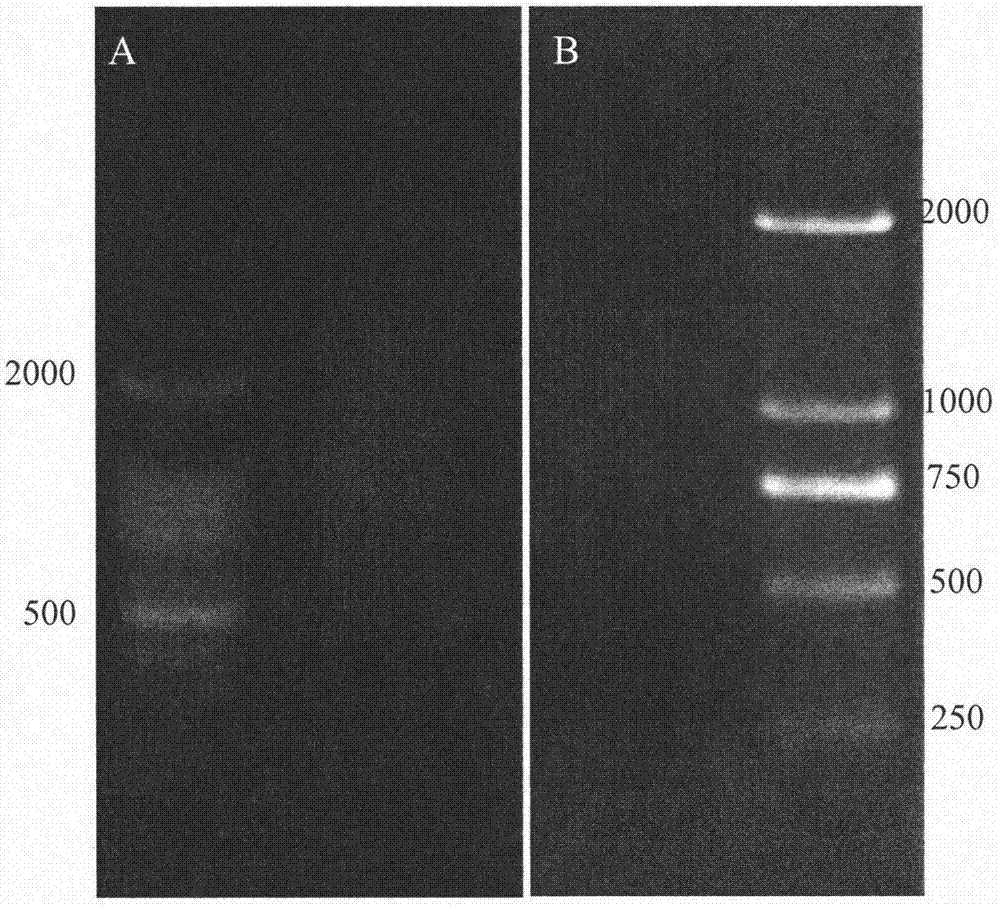
图2.scbs和mb-seq的文库扩增电泳图。a:scbs-seq;b:mb-seq。marker大小用数字标注在旁,单位bp。
图3.scbs-seq与mb-seq测序数据的基因组覆盖度比较。实线、虚线、点分别代表了scbs-seq、单细胞mb-seq、多细胞mb-seq的数据结果。
具体实施方式
下面将结合本发明中的实施例和附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
本发明所采取的mb-seq测序方法的操作流程如附图1所示,具体操作过程如下:
1.将玉米的四分体小孢子放入27%的甘露醇溶液,胞内渗透压平衡维持了正常的细胞形态。利用微型玻管、显微注射器、显微镜,分离单个细胞或细胞核于4μl纯水中。分离后将样品置于冰上待所有样品分离完毕进行下步或可移至-80℃冰箱备用;
2.配置8μl细胞裂解缓冲液(15mmtris-clph7.4,0.9%sdsand0.5μlproteinasek),将缓冲液加入4μl样品中,37℃孵育1小时,反应完后可执行下一步或移至-20℃备用;
3.使用imprintdnamodificationkit将未甲基化的胞嘧啶转化成尿嘧啶:
a)在上述裂解的dna中加入1μlbalancesolution,37℃孵育15分钟;
b)在dnamodificationpowder中加入1.1mldnamodificationsolution,涡旋2分钟待液体澄清后(若不澄清,可65℃水浴1分钟至固体全部溶解),加入40μlbalancesolution,涡旋混匀液体;
c)在dna样品中加入上步配置的试剂62.5μl;
d)65℃孵育1.5小时;
e)95℃孵育3分钟;
f)65℃孵育20分钟;
g)4℃孵育1分钟;
4.使用
a)将300μlbindingbuffer加入吸附柱中;
b)将1μlcarrierrna(epitectbisulfitekit)加入待纯化的dna样品,涡旋后加入吸附柱中;
c)将50μlbindingbuffer加入盛dna的pcr管中,转移残留的dna到吸附柱中;
d)涡旋吸附柱,6000rpm离心30秒,弃液;
e)在吸附柱中加入500μlwashbuffer,6000rpm离心30秒,弃液;
f)在吸附柱中加入100μl澄清的bdbuffer(epitectbisulfitekit),室温下静置15分钟,6000rpm离心30秒,弃液;
g)在吸附柱中加入300μlwashbuffer,6000rpm离心30秒,弃液;
h)再重复一遍上述操作;
i)13000rpm离心1分钟,将吸附柱插入到新的离心管中;
j)在吸附柱中加入20μleb,室温下静置5分钟,6000rpm离心30秒,转移清液于八连pcr管中,可放于-20℃备用;
5.短片段富集合成反应:
a)配置以下mix,混匀后加入八连pcr管中;
i.2.5μl10xnebuffer2;
ii1μl10mmdntpmix;
iii.1μl10umn9primar;
b)65℃反应3分钟,转移至冰上1分钟;
c)加入1ulklenowfragment;
d)4℃孵育5分钟,每15秒上升1℃,直到37℃;在37℃孵育30分钟;
e)4℃孵育4分钟,95℃孵育45秒,置于冰上退火成单链;
f)将以下试剂混匀,加入到样品中,再混匀;
i.0.1μl10mmdntp;
ii.0.25μl10xnebuffer2;
iii.1μl10umn9primer;
iv.0.5μlklenowfragment;
v.0.65μl纯水;
g)再重复步骤d到f三次(一共四次);
h)4℃孵育5分钟,每15秒上升1℃,直到37℃;在37℃孵育90分钟;4℃静置10分钟(一共扩增了五次);
6.磁珠纯化并去除单链引物:
a)在上一步产物中加入30μl(0.9倍体积)ampurexpbeads磁珠;
b)涡旋混匀后室温孵育10分钟;
c)置于磁铁上5分钟,移除上清液;
d)用200μl80%乙醇洗4遍,液流不直接冲刷磁珠,每次30秒;
e)室温下干燥,至磁珠表面有少许裂痕且管壁上没有小水珠;
f)移除磁铁,对准磁珠加入11.5μleb,混匀后室温放置2分钟;
g)再置于磁铁上5分钟;
h)吸出清液10μl进行酶连反应,也可暂时置于-20℃备用;
7.酶连反应:
a)将上述已纯化的10μl样品95℃孵育45秒,迅速置于冰上1分钟,至dna双链变性为单链;
b)混合以下试剂,混匀后加入上步样品中;
i.2.35μl纯水;
ii.1μlt4rnaligase1;
iii.1.5μlt410xbuffer;
iv.0.15μl100mmatp;
c)混匀后,37℃孵育30分钟;
8.mda扩增:
a)混合以下试剂,混匀后加入上步样品中;
i.5μl10xphi29buffer;
ii.10μl100μmbn9primer;
iii.14.5μl纯水;
iv.2.5μl10mmdntp;
v.1μl100xbsa;
vi.2μlphi29polymerase;
b)混匀后,30℃孵育2.5小时;
c)65℃孵育10分钟失活酶;
d)4℃保持1分钟,备用;
9.磁珠纯化:
a)在上一步产物中加入45μl(0.9倍体积)ampurexpbeads磁珠;
b)涡旋混匀后室温孵育10分钟;
c)置于磁铁上5分钟,移除上清液;
d)用200μl80%乙醇洗2遍,液流不直接冲刷磁珠,每次30秒;
e)室温下干燥,至磁珠表面有少许裂痕且管壁上没有小水珠;
f)移除磁铁,对准磁珠加入13μleb,混匀后室温放置2分钟;
g)再置于磁铁上5分钟;
h)吸出清液12μl,备用;
10.用qubit测定扩增产物的浓度:在199μlbuffer中加1μl染料,混匀后吸出1μl,加入1μl上步清液,混匀后在黑暗处静置2分钟,用qubit测定浓度;
11.基于tn5的dna建库试剂盒(trueprepdnalibraryprepkitv2for
a)将浓度调到5ng/11μl水中;
b)样品中加入以下试剂,涡旋混匀后55℃孵育10分钟,在保持于10℃中备用;
i.4μl5xttbl;
ii5μlttemixv5;
c)加入5μl5xts,混匀终止反应;
d)混合以下试剂,混匀后加入上步样品中;
i.4μl纯水;
ii.10μl5xtab;
iii.5μlindexn5;
iv.5μlindexn7;
v.1μltae;
e)混匀后,进行pcr富集反应:
i.72℃3分钟热启动;
ii.98℃30秒变性dna;
iii.98℃15秒变性;
iv.60℃30秒退火引物结合;
v.72℃3分钟模板链延伸;
vi.返回步骤iii九次(共十个循环);
vii.72℃5分钟酶失活;
viii.4℃保持1分钟,备用;
12.文库纯化与片段分选:
a)在上一步50μl产物中加入30μl(0.6倍体积)ampurexpbeads磁珠;
b)涡旋混匀后室温孵育5分钟;
c)置于磁铁上5分钟;
d)吸取上清液至另一pcr管中,在清液中加入7.5μlampurexpbeads磁珠;
e)涡旋混匀后室温孵育5分钟;
f)置于磁铁上5分钟,弃清液;
g)用200μl80%乙醇洗2遍,液流不直接冲刷磁珠,每次30秒;
h)室温下干燥,至磁珠表面有少许裂痕且管壁上没有小水珠;
i)移除磁铁,对准磁珠加入12μleb,混匀后室温放置2分钟;
j)再置于磁铁上5分钟;
k)吸出清液10μl,此为成品文库,可将其保存于-20℃备用;
13.上机测序:文库在用qpcr定量后,用illuminahiseq3000上机高通量测序。
对比例1
按照smallwood等提出的单细胞全基因组重亚硫酸盐测序文库(scbs)的构建方法(对照)(smallwoodsa,leehj,angermuellerc,etal.,single-cellgenome-widebisulfitesequencingforassessingepigeneticheterogeneity[j].natmethods,2014.11(8):817-820.),重点从重亚硫酸盐转换方法以及转换后小片段的扩增方式设置构建测序文库方法的对比试验。各组的处理方式如表1所示。
表1.测序文库不同构建方法的比较
试剂盒1为zymoezdnamethylation-goldtmkit(zymo,d5005),试剂盒2为sigmaimprintdnamodificationkit(sigma,mod50-1kt)。
对照组分别采用两种试剂盒进行重亚硫酸盐转换,转化后的小片段进行第一链第二链合成扩增,在玉米四分体小孢子中,scbs文库的片段大小从500bp到5kb不等,其文库的覆盖度较低,从而影响测序的覆盖度和比对率。为此我们希望利用mda这种扩增效率及测序覆盖度更高的方法来改善所有物种中单细胞甲基化测序的分辨度。
试验组1,2,3和4中,要么是先对短片段进行mda(试验组1和4),要么是试图先将低浓度的dna片段酶连(试验组2和3)。理论上,经过重亚硫酸盐处理的片段中未甲基化的c会转变成u,所以测序数据中gc%应较低。但四组试验产生的片段gc含量过高,平均为48.7%,不仅高于scbs数据的35%,还高于基因组的46%,可见上述四个实验组的方法不适于玉米单细胞甲基化测序文库的构建。
在试验组5中,先对重亚硫酸盐转化后的小片段进行富集,累计到一定浓度后,将dna变性成单链,酶连成长片段后再进行mda,扩增产物再利用tn5进行建库并上机测序。该方法测序数据的gc%平均为34%,与scbs的32%相近。
此外,在同等数据量时(试验5平均数据量为146m;scbs-seq平均数据量为103m),试验组5的方法平均序列比对率和覆盖度均明显高于scbs-seq。前者的序列比对率和覆盖度20.6%和17.0%,后者为3.5%和1.8%。因此,实验组5的方法五优于scbs-seq方法。试验5的方法即本发明的mb-seq。
同时,如附图2所示,电泳检测玉米单细胞中scbs文库和mb-seq文库的片段大小分别为500bp-5kb和500bp左右。mb-seq文库片段大小更符合hiseq测序要求的300-600bp,能包含更大的文库丰富度,从而提高测序的覆盖度。
进一步地,如附图3所示,从各测序材料中mappedreads数与基因组覆盖度之间的关系可以看出,ms-seq的可重复度高,稳定性好。
还需要说明的是,在可实施且不明显违背本发明的主旨的前提下,在本说明书中作为某一技术方案的构成部分所描述的任一技术特征或技术特征的组合同样也可以适用于其它技术方案;并且,在可实施且不明显违背本发明的主旨的前提下,作为不同技术方案的构成部分所描述的技术特征之间也可以以任意方式进行组合,来构成其它技术方案。本发明也包含在上述情况下通过组合而得到的技术方案,并且这些技术方案相当于记载在本说明书中。
上述说明展示并描述了本发明的优选实施例,如前所述,应当理解本发明并非局限于本文所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本文所述发明构想范围内,通过上述教导或相关领域的技术或知识进行改动。故本发明包括但不限于上述实施方式,任何符合本权利要求书或说明书描述,符合与本文所公开的原理和新颖性、创造性特点的方法、工艺、产品,均落入本发明的保护范围之内。
工业实用性
根据本发明,能够适用于少量细胞样品及单细胞样本的重亚硫酸盐测序用文库的构建方法及建库试剂盒。
- 还没有人留言评论。精彩留言会获得点赞!