一种吡咯类衍生物的合成方法与流程
本发明涉及化工中间体领域,具体涉及多取代吡咯生物及其合成方法。
背景技术:
现有技术的缺点和不足(发明目的):多取代吡咯是一类重要的含氮杂环化合物,广泛存在于许多天然产物和高活性的药物中。该类杂环体系通常有良好的抗真菌、抗炎、抗惊厥等生物活性。
鉴于吡咯衍生物的重要性,它们在合成化学领域引起了极大的关注,并发展了许多不同的合成策略。除了经典的paal-knorr或hantazsch反应外,还有一些新的合成方法,包括1,3-偶极环加成,c-h活化,多组分反应,氧化偶联环化反应等。
例如,毕锡和与雷爱文课题组同时报道了通过异氰化物与未活化的末端炔烃的银催化环加成反应,制备了一种通用而实用的吡咯构筑方法(angew.chem.int.ed.,2013,52,6953-6957;angew.chem.int.ed.,2013,52,6958-6961.)。
pagadala描述了一种由芳醛、丙二腈、异氰化物和环状仲胺无催化剂的四组分反应生成具有抗肿瘤活性的官能化吡咯的有效方法(org.biomol.chem.,2015,13,1800-1806.)。
2019年,li报道了一种钯催化的α-对甲苯磺酰胺联烯的氧化反应,用于一锅法合成吡咯环(acscatal.,2019,9,5184-5190.)。虽然文献中关于吡咯的合成有许多优点,但我们不能忽视这些方法往往伴随着一些缺陷,如反应温度过高,需要过酸或者过碱的环境,添加了有毒的添加剂,操作复杂等等。因此,新的、简便的吡咯合成方法在药物化学和合成化学中仍具有重要的应用价值。
技术实现要素:
本发明是提供一种吡咯类化合物的合成方法,利用简单易得的原料,通过一锅法合成一类结构新颖的多取代吡咯类化合物。
本发明的技术方案是:
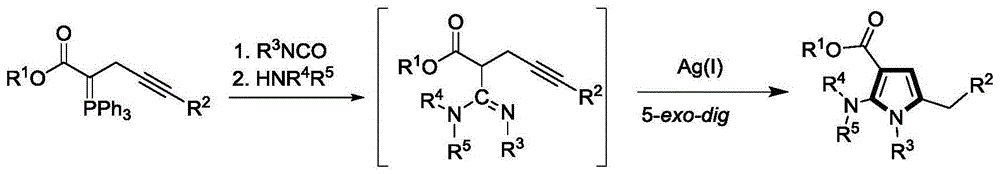
一种吡咯类衍生物的合成方法,包括步骤:在装有膦叶立德的容器中加入合适的反应溶剂,依次加入异氰酸酯和胺类化合物,在常温下搅拌,随后加入硝酸银,继续搅拌直至反应结束、减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物吡咯类衍生物,其反应式如下:
其中,
r1是甲基、乙基的任意一种;
r2:氢、甲基的任意一种;
r3:苯基、4-甲基苯基、3-甲基苯基、3,5-二甲基苯基、4-甲氧基苯基、4-氟苯基、4-氯苯基、2-氯苯基的任意一种。
nr4r5:哌啶基、二乙胺基、四氢吡咯烷基、吗啡琳基、二苄胺基、n-甲基苄胺基、叔丁氨基、正丁胺基、苄胺基、异丙胺基的任意一种;
上述的反应温度是常温;
膦叶立德化合物、异氰酸酯化合物、胺类化合物之间的摩尔比为1:1:1。
进一步地,上述的异氰酸酯是选自苯基异氰酸酯、4-甲基苯基异氰酸酯、3-甲基苯基异氰酸酯、3,5-二甲基苯基异氰酸酯、4-甲氧基苯基异氰酸酯、4-氟苯基异氰酸酯、4-氯苯基异氰酸酯、2-氯苯基异氰酸酯的任意一种。
胺可以是选自哌啶、二乙胺、四氢吡咯烷、吗啡琳、二苄胺、n-甲基苄胺、叔丁氨、正丁胺、苄胺、异丙胺的任意一种。
进一步地,上述的反应溶剂是选自二氯甲烷、四氢呋喃中的任意一种。
进一步地,上述的膦叶立德的制备方法:将溴乙酸酯基叶立德1溶于三氯甲烷中,加入溴丙炔2,设置60℃搅拌回流过夜;然后冷却反应体系,旋干溶剂,加入大量水,滴加氢氧化钠溶液调节至碱性并搅拌反应体系5分钟;用二氯甲烷萃取,有机相用无水硫酸钠干燥后,滤去硫酸钠,得有机相,浓缩有机相得到原料膦叶立德;
一种吡咯类衍生物的合成方法,在圆底烧瓶中加入原料叶立德以及溶剂一,随后加入异氰酸酯,常温下搅拌反应4-6小时得到中间体4,不经分离随后往体系中直接加入胺继续搅拌1-2小时得到脒中间体5,旋干溶剂,往体系中加入溶剂二,加入催化剂硝酸银,常温搅拌12-24小时,经tlc监测反应反应完成后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离得到目标产物;
进一步地,上述溶剂一为二氯甲烷;溶剂二为四氢呋喃;所述的膦叶立德化合物、异氰酸酯化合物、胺类化合物之间的摩尔比为1:1:1。
2-(二乙氨基)-1-(4-甲氧基苯基)-5-甲基-1h-吡咯-3-羧酸乙酯的合成进一步地,上述原料为叶立德3b,加入物为甲氧基苯基异氰酸酯,常温下搅拌4小时,不经分离随后往体系中直接加入二乙胺继续搅拌1小时,常温搅拌12小时;
叶立德3b结构式为:
与现有技术相比,本发明的有益效果如下:
本发明提供了一种一锅法制备多取代吡咯类化合物的方法,通过简单的原料开始,反应在常温下即可进行,一锅法的制备方法省去了中间体分离的步骤,使得操作更加简便,具有操作简便、条件温和、底物适用范围广、原料廉价易得、成本低,目标产物产率。
附图说明
图1是本发明的多取代吡咯类化合物的的化学反应式;
图2是2-(二乙氨基)-1-(3,5-二甲基苯基)-5-甲基-1h-吡咯-3-羧酸甲酯的的1hnmr图;
图3是2-(二乙氨基)-1-(3,5-二甲基苯基)-5-甲基-1h-吡咯-3-羧酸甲酯的的13cnmr图。
具体实施方式
下面结合附图对于本发明作进一步的说明:
吡咯类衍生物的合成方法,其特征在于,包括步骤:
在装有膦叶立德的容器中加入合适的反应溶剂,依次加入异氰酸酯和胺类化合物,在常温下搅拌,随后加入硝酸银,继续搅拌直至反应结束、减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物吡咯类衍生物,其反应式如下:
其中:反应溶剂是选自二氯甲烷、四氢呋喃中的任意一种;
反应温度可以是常温;
膦叶立德化合物、异氰酸酯化合物、胺类化合物之间的摩尔比为1:1:1;
异氰酸酯可以人是选自苯基异氰酸酯、4-甲基苯基异氰酸酯、3-甲基苯基异氰酸酯、3,5-二甲基苯基异氰酸酯、4-甲氧基苯基异氰酸酯、4-氟苯基异氰酸酯、4-氯苯基异氰酸酯、2-氯苯基异氰酸酯的任意一种。
胺可以是选自哌啶、二乙胺、四氢吡咯烷、吗啡琳、二苄胺、n-甲基苄胺、叔丁氨、正丁胺、苄胺、异丙胺的任意一种。
实施例1,原料膦叶立德3的制备:参考文献((a)b.m.trost,g.d.probstanda.schoop,j.am.chem.soc.1998,120,9228-9236.(b)d.scholz,s.weber-roth,e.macorattiande.francotte,syntheticcommunications,1999,29,1143-1155.(c)c.zhu,b.yang,t.jiangandj.e.backvall,angew.chem.int.ed.,2015,54,9066-9069.(d)n.britto,v.g.gore,r.s.malianda.c.ranade,syntheticcommunications,1989,19,1899-1910.),将乙酸酯基叶立德1(10mmol)溶于三氯甲烷中,加入溴丙炔2(10mmol),设置60℃搅拌回流过夜。然后冷却反应体系,旋干溶剂,加入大量水,滴加氢氧化钠溶液调节至碱性并搅拌反应体系5分钟。用二氯甲烷萃取,有机相用无水硫酸钠干燥后,滤去硫酸钠,得有机相,浓缩有机相得到原料3,产率。
膦叶立德3a的制备:将2-(三苯基-λ5-亚膦基)乙酸甲酯(10mmol)溶于三氯甲烷中,加入3-溴丙炔(10mmol),设置60℃搅拌回流12小时。然后冷却反应体系,旋干溶剂,加入大量水,滴加氢氧化钠溶液调节至碱性并搅拌反应体系5分钟。用二氯甲烷萃取,有机相用无水硫酸钠干燥后,滤去硫酸钠,得到有机相,浓缩有机相得到2-(三苯基-λ5-亚磷酰基)戊基-4-乙酸甲酯3a,产率67%。
膦叶立德3b的制备:将2-(三苯基-λ5-亚膦基)乙酸乙酯(10mmol)溶于三氯甲烷中,加入3-溴丙炔(10mmol),设置60℃搅拌回流12小时。然后冷却反应体系,旋干溶剂,加入大量水,滴加氢氧化钠溶液调节至碱性并搅拌反应体系5分钟。用二氯甲烷萃取,有机相用无水硫酸钠干燥后,滤去硫酸钠,得到有机相,浓缩有机相得到2-(三苯基-λ5-亚膦酰基亚乙基)戊基-4-壬酸酯3b,产率68%。
膦叶立德3c的制备:将2-(三苯基-λ5-亚膦基)乙酸甲酯(10mmol)溶于三氯甲烷中,加入1-溴2-丁炔(10mmol),设置60℃搅拌回流12小时。然后冷却反应体系,旋干溶剂,加入大量水,滴加氢氧化钠溶液调节至碱性并搅拌反应体系5分钟。用二氯甲烷萃取,有机相用无水硫酸钠干燥后,滤去硫酸钠,得到有机相,浓缩有机相得到2-(三苯基-λ5-膦基亚甲基)己基-4-壬酸酯3c,产率65%。
其中膦叶立德3可分以下种结构式为:
实施例2,吡咯6的制备,如下图示反应式说明,具体实施方式:在50ml的圆底烧瓶中加入原料叶立德3(2mmol)以及10ml二氯甲烷,随后加入异氰酸酯(2mmol),常温下搅拌反应4-6小时得到中间体4,不经分离随后往体系中直接加入2mmol胺继续搅拌1-2小时得到脒中间体5,旋干溶剂,往体系中加入10ml四氢呋喃,硝酸银(0.2mmol),常温搅拌12-24小时,经tlc监测反应反应完成后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚=1:10)得到目标产物6。
实施案例3
5-甲基-1-苯基-2-(哌啶-1-基)-1h-吡咯-3-羧酸甲酯的合成
在反应容器中加入2mmol原料叶立德3a于10ml二氯甲烷中溶解,然后加入2mmol苯基异氰酸酯,常温下搅拌4小时,不经分离随后往体系中直接加入2mmol哌啶继续搅拌1小时,旋干溶剂,往体系中加入10ml四氢呋喃,硝酸银(0.2mmol),常温搅拌24小时,反应结束后减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物,产率83%。
实施案例4
2-(二乙氨基)-5-甲基-1-(对甲苯基)-1h-吡咯-3-羧酸甲酯的合成在反应容器中加入2mmol原料叶立德3a于10ml二氯甲烷中溶解,然后加入2mmol4-甲基苯基异氰酸酯,常温下搅拌4小时,不经分离随后往体系中直接加入2mmol二乙胺继续搅拌1小时,旋干溶剂,往体系中加入10ml四氢呋喃,硝酸银(0.2mmol),常温搅拌16小时,反应结束后减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物,产率86%。
实施案例5
5-甲基-2-(哌啶-1-基)-1-(间甲苯基)-1h-吡咯-3-羧酸甲酯
在反应容器中加入2mmol原料叶立德3a于10ml二氯甲烷中溶解,然后加入2mmol间甲基苯基异氰酸酯,常温下搅拌6小时,不经分离随后往体系中直接加入2mmol哌啶继续搅拌1小时,旋干溶剂,往体系中加入10ml四氢呋喃,硝酸银(0.2mmol),常温搅拌16小时,反应结束后减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物,产率86%。
实施案例6
2-(叔丁基氨基)-5-甲基-1-苯基-1h-吡咯-3-羧酸甲酯的合成
在反应容器中加入2mmol原料叶立德3a于10ml二氯甲烷中溶解,然后加入2mmol苯基异氰酸酯,常温下搅拌5小时,不经分离随后往体系中直接加入2mmol叔丁胺继续搅拌1小时,旋干溶剂,往体系中加入10ml四氢呋喃,硝酸银(0.2mmol),常温搅拌20小时,反应结束后减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物,产率74%。
实施案例7
2-(二乙氨基)-1-(4-甲氧基苯基)-5-甲基-1h-吡咯-3-羧酸乙酯的合成
在反应容器中加入2mmol原料叶立德3b于10ml二氯甲烷中溶解,然后加入2mmol对甲氧基苯基异氰酸酯,常温下搅拌4小时,不经分离随后往体系中直接加入2mmol二乙胺继续搅拌1小时,旋干溶剂,往体系中加入10ml四氢呋喃,硝酸银(0.2mmol),常温搅拌12小时,反应结束后减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物,产率90%。
实施案例8
2-(二乙氨基)-1-(3,5-二甲基苯基)-5-甲基-1h-吡咯-3-羧酸甲酯的合成
在反应容器中加入2mmol原料叶立德3b于10ml二氯甲烷中溶解,然后加入2mmol3,5-二甲基苯基异氰酸酯,常温下搅拌4小时,不经分离随后往体系中直接加入2mmol二乙胺继续搅拌1小时,旋干溶剂,往体系中加入10ml四氢呋喃,硝酸银(0.2mmol),常温搅拌18小时,反应结束后减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物,产率86%。
实施案例9
1-(4-氯苯基)-2-(二丙基氨基)-5-甲基-1h-吡咯-3-羧酸甲酯的合成
在反应容器中加入2mmol原料叶立德3a于10ml二氯甲烷中溶解,然后加入2mmol4-二氯苯基异氰酸酯,常温下搅拌6小时,不经分离随后往体系中直接加入2mmol二乙胺继续搅拌1小时,旋干溶剂,往体系中加入10ml四氢呋喃,硝酸银(0.2mmol),常温搅拌24小时,反应结束后减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物,产率69%。
实施案例10
2-(苄基(甲基)氨基)-5-乙基-1-(对甲苯基)-1h-吡咯-3-羧酸甲酯的合成
在反应容器中加入2mmol原料叶立德3c于10ml二氯甲烷中溶解,然后加入2mmol4-甲基苯基异氰酸酯,常温下搅拌4小时,不经分离随后往体系中直接加入2mmoln-甲基苄胺继续搅拌1小时,旋干溶剂,往体系中加入10ml四氢呋喃,硝酸银(0.2mmol),常温搅拌20小时,反应结束后减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得目标产物,产率76%。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!