用于确定高同源性区域中的基因型的方法与流程
以下公开内容总体上涉及确定基因型,并且更具体地,涉及确定与具有对应的高度同源性同源物的基因相关联的基因型。
技术背景
许多疾病源自因突变而失活的基因。因此,对此类突变的识别是临床基因医学的基本目标。对于许多基因而言,这些突变相对易于从下一代测序(ngs)数据中找到。然而,对于多种重要且普遍的病症的受试者的基因的子集而言,识别失活基因并计数其数量是具有挑战性的,因为这些基因被基因组的其他同源部分有效地遮蔽。
解析与其他(通常功能失调的)区域高度同源的基因组区域的结构和内容是特别困难的,甚至在使用先进ngs工具的情况下同样如此。不幸的是,这些技术障碍是尤其成问题的,因为许多这些困难区域具有疾病暗示。实际上,它们与功能失调区域的极大同源性导致基因与同源物之间频繁的重排,这可能影响基因的功能性拷贝的数量。
因此,仍然需要检测并确定受试者关于基因的基因型和/或携带状态,其中所述基因具有同源性同源物。
发明概述
允许确定高度同源性基因和对应同源物的基因型的当前技术是时间密集型和劳动密集型的,并且是昂贵的,从而使得它们不适于广泛的临床使用。
目前公开的方法可以可承受的且高通量的方式进行实践。因此,节省了大量的时间、劳力和花费。此外,本发明方法解决解析在以下区域中的结构/拷贝数/基因型的问题,在所述区域中ngs读取与基因或其同源物的唯一比对受到损害。重要的是,这些损害的“高度同源性”区域是基于两个特征:(1)给定的实验中的ngs读取的长度,以及(2)由比对软件(例如bwa)所允许的错配的量。
在一方面,本文提供一种用于确定个体关于感兴趣基因的基因组结构(即,基因型)的方法,其中感兴趣基因具有高度同源性同源物。
在一个实施方案中,感兴趣基因以及其同源物的序列信息使用针对外显子的引物。在某些实施方案中,所述序列信息来自感兴趣基因和/或同源物的内含子。在某些实施方案中,所述序列信息来自基因间区。
在另一实施方案中,所述序列信息由下一代测序(ngs)生成。在一些实施方案中,ngs是高深度全基因组鸟枪测序(即,不使用富集探针)。在其他实施方案中,ngs是靶向测序,例如像杂交捕获技术、多重扩增子富集、或使用于测序反应的基因组特定区域富集的任何其他手段。在一些实施方案中,所述测序以多重测定进行。
在一个实施方案中,基因是smn1并且假基因是smn2。在一个实施方案中,改变的拷贝数的smn1的存在指示受试者可能是疾病脊髓性肌萎缩(sma)的携带者。
在另一个实施方案中,基因是cyp21a2并且假基因是cyp21a1p。在一个实施方案中,改变的拷贝数的cyp21a2的存在指示受试者可能是疾病先天性肾上腺皮质增生症(cah)的携带者。
在一个实施方案中,基因是hba1并且同源物是hba2(或反之亦然)。在一个实施方案中,改变的拷贝数的hba1或hba2的存在指示受试者可能是疾病α地中海贫血的携带者。
在另一实施方案中,基因是gba并且假基因是gbap。在一个实施方案中,改变的拷贝数的gba的存在指示受试者可能是疾病戈谢氏病(gaucher’sdisease)的携带者。
在一个实施方案中,基因是pms2并且假基因是pms2cl或多种其他假基因中的一种。截止2015年12月,存在15种假基因。假基因可选自但不限于称为pms2cl的13种假基因,其中13种假基因中其他12种编号为pms2p1到pms2p12。在一个实施方案中,改变的拷贝数和/或改变基因和假基因的取向的倒位(例如,使假基因的部分与基因融合并且因此损害基因功能的那些倒位)的存在可指示受试者具有增加的患疾病林奇综合征(lynchsyndrome)的风险。
在一个实施方案中,基因是chek2,其具有多种假基因。截止2014年12月,存在七种假基因。所述假基因可选自但不限于在经过验证的数据库(curateddatabase)中所列举的chek2假基因。在一个实施方案中,通过与其假基因重组产生的突变(例如,假基因衍生的移码突变)的存在可指示受试者具有增加的患疾病乳腺癌以及其他疾病的风险。在本领域中熟知的是,七种假基因中仅一种已经被命名,并且风险主要与一种突变1100delc相关联。然而,其他突变也导致患疾病的风险。患者处于患李-佛美尼综合症(lifraumenisyndrome)和其他可遗传癌症的风险下。
在一方面,提供一种配置来执行用于实施本文所述的方法的指令的计算机系统。
通过以下详细描述,本发明的其他目的、特征和优势将变得显而易见。然而,应理解,尽管指示本发明的优选实施方案,但是详细描述和特定实施例仅通过说明的方式给出,因为在本发明的范围和精神内的各种改变和修改对于通读此详细描述的本领域技术人员而言将变得显而易见。
附图简述
图1示出基因以及其同源物(例如,在假基因的情况下是功能失调的同源物)的各种基因组结构。在“正常的”样品中,基因以及其同源物各自有两个拷贝。对于许多具有同源物的基因而言(实际上,对于引起戈谢氏病、脊髓性肌萎缩(“sma”)、先天性肾上腺皮质增生症(“cah”)和α地中海贫血的基因以及与各种癌症相关的多种基因而言),基因和同源物在染色体上彼此相对接近。示出经历基因和/或同源物的“缺失或复制”的染色体的一些实例。基因与同源物之间的重组可产生“融合”基因,所述“融合”基因一部分是“基因”并且一部分是“同源物”。最后,基因与同源物之间的序列的“互换”是相对频繁的。
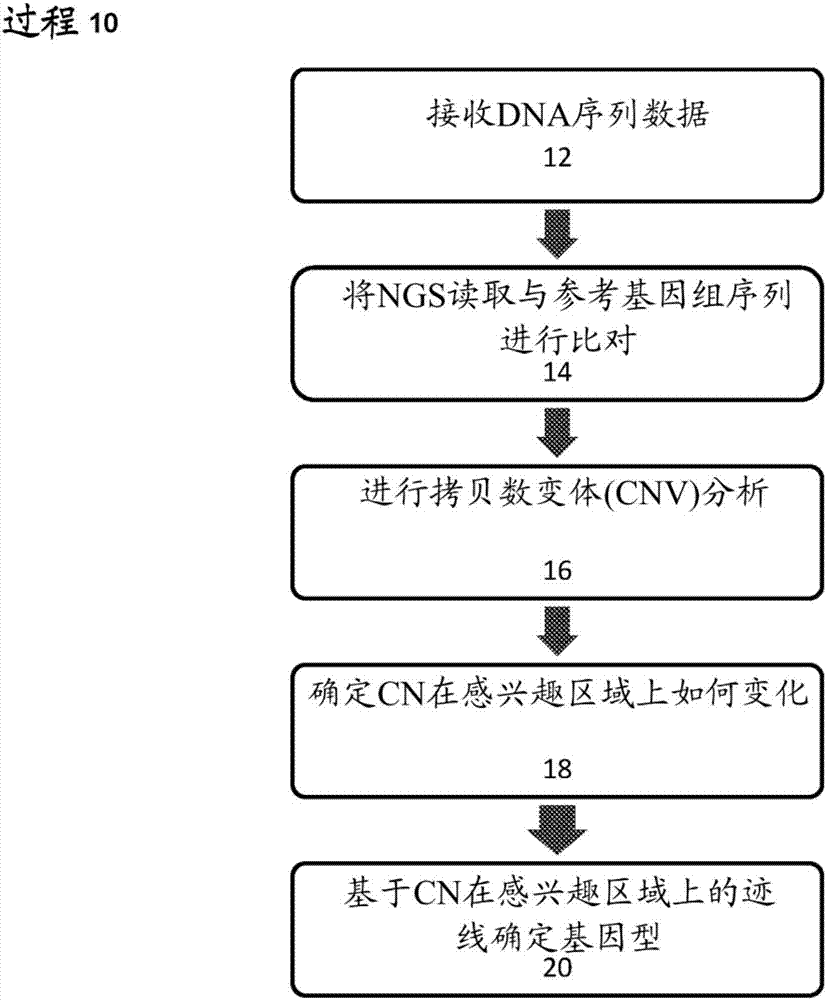
图2是如本文所述的方法的流程图。
图3示出本发明的各种实施方案可在其中运行的示例性系统和环境。
图4示出示例性计算系统。
图5是smn1和smn2的拷贝数(“cn”)图。对于10,000个样品而言,使用本文所述的测序数据和cn分析来计算样品的smn1和smn2的cn,并且然后使用这些值分别作为散点图中的x坐标和y坐标。每个样品的cn(smn1)(即,smn1的拷贝数)通过基于正交qpcr的测定来验证:通过此后一种测定确定具有1个、2个或3个拷贝的样品分别由圆圈、三角形和正方形指示。应注意,在具有cn(smn1)=1和cn(smn1)=2的点之间的测序数据中有非常清楚的间隔。实际上,使用cn(smn1)=1.4的截取值来将样品分类为具有1个或2个smn1拷贝,基于测序的cn分析将不产生假阳性或假阴性。其他值得注意的特征或曲线包括:(1)最高点密度在(2,2)附近,这是轨迹的正常排列;(2)然而,许多样品距离(2,2)很远,和smn1与smn2之间频繁的转换/缺失/复制一致。
图6示出gba和gbap的两个拷贝数图。对于两个单一患者样品,在九个不同位点处绘制gba以及其同源物/假基因gbap的cn值,从5’到3’(左到右)排列。顶部样品(a)是正常的,因为它具有gba和gbap两者的两个拷贝。然而,底部样品(b)经历“互换”事件,其中gbap拷贝之一的3’端获得gba衍生的序列。
图7是hba1和hba2的拷贝数图。所述曲线图示出48个患者样品在围绕并包括hba2和hba1的区域中的cn值。粗线示出单个染色体的较大区段已缺失的单个样品,因此其在图的右侧的大部分信号下降。正如所预期的,大部分样品具有cn=2。三个样品具有在z1与z2区域之间发生的短缺失。
图8是示出用于cyp21a2基因以及其同源物cyp21a1p中的每个探针的拷贝数的图。所述曲线图示出48个患者样品在基因cyp21a2(a;左)(其影响cah)以及其假基因cyp21a1p(b;右)中的cn值。x轴上的每个位置是基因中的不同位点,从5’到3’排列。三个粗迹线是已知经历使基因拷贝之一消除的融合事件的样品,因此在左边的基因曲线图中它们的cn值是~1和~0。cyp21a2和cyp21a1p在整个演变过程中经历相当多的互换/融合/缺失/复制,这是为何它们在以上曲线图中的迹线比先前图中针对戈谢氏病(图6)和α地中海贫血(图7)的cn迹线更参差不齐。应注意,本文所述的cn分析方法的关键目标之一是想要确定功能性基因拷贝(即,在此情况下是cyp21a2)的数量。由此,首先看到靠近5’端的位点并且使用它们的平均值来分解cn(cyp21a2)。接下来,考虑整个迹线(即,包括3’端)以确定所发生的重排列的类型。
图9是示出样品数据如何从原始读取计数处理成可针对拷贝数变化解读的值的图。示出了在本文下文中进一步描述的六个步骤和五个示例性表(标示为a、b、c、d和e)。
此专利的文件包含至少一幅彩色绘图。在提出请求并支付必要费用后,本事务所将提供具有彩色绘图的本专利或专利出版物的副本。
详细描述
现在将使用以下定义和实施例仅以参考的方式对本发明进行详细描述。本文中所提及的所有专利和出版物(包括在此类专利和出版物内所公开的所有序列)明确地以引用的方式并入。
除非在本文中另外定义,否则本文所使用的所有技术和科学术语均具有与本发明所属领域的普通技术人员通常所理解的相同的意思。singleton等,dictionaryofmicrobiologyandmolecularbiology,第2版,johnwiley和sons,newyork(1994),以及hale和marham,theharpercollinsdictionaryofbiology,harperperennial,ny(1991)为本领域技术人员提供用于本发明中的许多术语的一般词典。虽然在本发明的实践或测试中可使用类似或等同于本文所描述的那些方法和材料的任何方法和材料,但是描述优选的方法和材料。对于本领域的定义和术语,从业者可具体参考sambrook等,1989和ausubelfm等,1993。应理解,本发明不限于所描述的具体方法、方案和试剂,因为这些方法、方案和试剂可变化。
数字范围包括限定所述范围的数字。术语“约”在本文中用于意指加上或减去数值的百分之十(10%)。例如,“约100”是指90与110之间的任何数字。
除非另有指出,否则分别地核酸从左至右以5'至3'取向书写;氨基酸序列从左至右以氨基至羧基取向书写。
本文所提供的标题不是对本发明的各种方面或实施方案的限制,这些方面或实施方案可通过参考整个说明书来获得。因此,即将在下文中定义的术语通过参考整个说明书而得到更充分地定义。
定义
如本文所用的,“纯化”意指分子以包含所述分子的样品的至少95重量%或至少98重量%的浓度存在于样品中。
“分离”分子是与例如在其天然环境下通常与它缔合的至少一种其他分子分开的核酸分子。分离的核酸分子包括在通常表达核酸分子的细胞中所含有的核酸分子,但是核酸分子存在于染色体外或存在于不同于其天然染色体位置的染色体位置处。
术语“%同源性”在本文中与本文中的术语“%同一性”互换使用,并且是指当使用序列比对程序进行比对时,编码发明性多肽或发明性多肽的氨基酸序列中的任一个的核酸序列之间的核酸或氨基酸序列同一性水平。在核酸的情况下,所述术语还适用于内含子和/或基因间区域。
例如,如本文所用的,80%同源性意指相同的事物为通过定义的算法确定的80%序列同一性,并且因此给定序列的同源物在给定序列的长度上具有大于80%的序列同一性。序列同一性的示例性水平包括但不限于对给定序列,例如如本文所述的发明性多肽中任一种的编码序列的80%、85%、90%、95%、98%或更大序列同一性。
可用于确定两个序列之间的同一性的示例性计算机程序包括但不限于blast程序组,例如可在因特网上公开获得的blastn、blastx和tblastx、blastp和tblastn以及blat。也参见altschul等,1990和altschul等,1997。
当相对于基因库dna序列和其他公开数据库中的核酸序列评估给定的核酸序列时,通常使用blastn程序来实施序列查找。blastx程序优选用于查找已经在针对基因库蛋白序列和其他公开数据库中的氨基酸序列的所有阅读框中翻译的核酸序列。blastn和blastx两者均使用默认参数11.0的开放缺口罚分(opengappenalty)和1.0的延伸缺口罚分(extendedgappenalty)来运行,并利用blosum-62矩阵。(参见,例如altschul,s.f.等,nucleicacidsres.25:3389-3402,1997。)
为了确定两个或更多个序列之间的“%同一性”而进行的所选序列的优选比对使用例如以默认参数操作的macvector版本13.0.7的clustal-w程序以及blosum30相似性矩阵来进行,所述默认参数包括10.0的开放缺口罚分、0.1的延伸缺口罚分。
如本文所用的,“高度同源的”意指基因与其对应的同源物之间的同源性在其长度对应于ngs读取长度的区域上大于90%。因此,如果基因中的任何区域与同源物是高度同源的,那么基因以及其同源物就称为是“高度同源的”。ngs读取长度的范围可以是30nt至400nt、50nt至250nt、50nt至150nt或100nt至200nt。重要的是,基因具有同源物,并不需要整个基因的序列是“高度同源的”;仅需要基因中的一个区域是高度同源的。
如本文所用的术语“同源物”是指与位于受试者基因组中其他位置处的感兴趣基因相同或几乎相同的dna序列。同源物可以是另一个基因、“假基因”或不是基因的一部分的序列区段。
如本文所用的术语“突变”是指自生的和遗传的序列变异,包括但不限于个体之间的变异或个体的序列与参考序列之间的变异。示例性突变包括但不限于snp、插入缺失(indel)、拷贝数变体、倒位、易位、染色体融合等。
如本文所用的“假基因”是非常类似于dna序列中的基因但是具有使其功能失调的至少一个变化的dna序列。所述变化可以是单个残基突变。所述变化可产生剪接变体。所述变化可导致翻译提前终止。假基因是功能性基因的功能失调相关基因。假基因的特征在于对已知基因(即,感兴趣基因)的同源性和非功能性的组合。
基因的假基因数量不限于本文中所列举的那些。假基因得到逐渐增加的认识。因此,本领域的技术人员将能够基于序列同源性或通过参考经过验证的基因库例如像基因卡(genecards.org)、pseudogenes.org等来确定序列是否是假基因。
如本文所用的,“感兴趣基因”是需要确定功能性拷贝的数量的基因。通常,由于两个染色体各自具有感兴趣基因的拷贝,所以感兴趣基因具有两个功能性拷贝。术语“感兴趣基因”和“基因”可在本文中互换使用。
过程
在可能的情况下,将来自感兴趣区域中的序列与杂交捕获探针或pcr引物富集,所述杂交捕获探针或pcr引物应被设计来使得所捕获和所测序的片段包含辨别基因与其同源物的至少一个序列。例如,杂交捕获探针可被设计来与在基因与同源物/假基因之间不同的几个碱基(“差别碱基(diffbases)”)相邻地退火。在缺少此类辨别序列的情况下,应使用多个探针来捕获可辨别片段,以便减少每个特定探针的序列所固有的偏差影响。扩增子测序可用作杂交捕获的替代方案,以作为实现靶向测序的手段。高深度全基因组测序可用作靶向测序的替代方案。可使用反映特定基因组区域的剂量的任何高通量定量数据,无论它来自ngs、微阵列或任何其他高通量定量分子生物学技术。
具有基因衍生的或同源物衍生的碱基的ngs序列读取的丰度允许区分正常个体(cn=2)与变种个体(cn≠2)。然而,另外的有用信息可甚至从不能辨别基因与同源物的序列读取获得,如在hba1和hba2的情况下,其中两个相同基因的正常组合cn是4,并且任一基因的缺失导致合计cn≤3。应注意,原则上,本文所述的cn分析可甚至应用于高深度全基因组鸟枪测序(即,不使用富集探针)。
广义地说,并且在一个实施例中,为了产生对区域的调用,进行以下过程,所述过程在图2中示出为过程10。首先,在12处获得感兴趣序列。例如,读取可以任何方式从与调用区域重叠(或者严格地说在其同源物的区域中)的bam文件中收集。这些读取然后可使用其相关软限幅信息来限幅。然后可舍弃来自对准器的辅助信息,例如碱基到碱基的比对信息,并且读取简单地变为碱基的序列。(在一些实施例中,可任选地进行基于映射质量的过滤。)
基于辨别基因与同源物的碱基的存在,对基因或同源物的读取进行区分。在此区分过程中所采用的辨别碱基取决于感兴趣的特定基因。另外,区分可再次基于特定应用,仅使用给定读取中的辨别碱基的子集。在杂交捕获探针序列自身成为所测序片段的一部分的实施方案中,所述杂交捕获探针被设计成使得辨别碱基是在末端配对读取的各端之一的末端处或末端附近。例如在此情况下,杂交捕获探针长度是例如39个碱基,但是测序仪从捕获的片段读取了40个碱基。探针被设计成使得第40个碱基是辨别碱基,从而允许整个读取(即,末端配对读取的两个端)基于第40个位置的碱基区分为基因或同源物。以上实施例中的精确数字(即,39和40)可改变并产生类似的结果。原则上,探针可短至10bp或长至1000bp,虽然20bp-100bp的长度范围是最常见的。在类似于以上实施方案且探针成为所测序片段的一部分的实施方案中,测序仪读取的长度必须超过探针长度至少1bp;然而,在单独的捕获片段包含足够将读取适当地区分成基因或同源物的辨别碱基的实施方案中,那么测序不一定需要延伸超过探针的长度。
在图9中示出实验数据的示例性处理。示出了来自具有根据单个实验(使用一个illumina流动池)的数据的表的摘录。每一排是一个样品。通常,在单个实验中处理(即,测试)48个或96个样品(即,“样品x”=“样品96”),虽然所述分析对于更多或更少样品均是有效的。所述分析强有力地利用了以下事实:拷贝数突变是相对少见的,尤其是在与疾病相关的基因中;因此,预期大部分样品将在每个位点处具有野生型拷贝数(“cn”)(即,cn=2)。
如图9表a所示,位点可区分成测试位点(例如,“ts1”、“ts2”等)和对照位点(例如,“cs1”、“cs2”等)。测试位点(ts)相对于对照位点(cs)的解析取决于以下测定:例如,在戈谢氏病测定中,ts是gba或gbap中的位点,并且cs包括基因组中具有不在gba或gbap中的数据的任何位点。作为另一个实施例,对于sma测试,仅有两个ts位点(一个针对smn1并且另一个针对smn2)。通常,对于每个实验,有数百个cs。如果独立进行cn分析,那么应使用至少10个cs,其中50个或更多个是优选的(基本地,需要足够的位点来得到对中间值的稳健测量,将在图9表b中看到这一点)。
下一步骤在图9表b中进行描绘,其中对cs原始读取值的中间值进行计算。应注意,表中的每个单元均可含有整数值的原始读取或调整读取的浮点数,其中读取数的调整可将类似于由于gc含量而引起的测序偏差的因素考虑在内。应注意,这仅涉及cs,因为初始假设是这些值具有cn=2;如果给定排的样品具有cn突变并且ts的数量超过cs,那么此时包括ts可能使所述排的中间值偏斜。与使用表示平均值的均数不同,中间值对于离群读取值是稳健的,所述离群读取值在测序数据中是普遍的;然而,仍然应具有至少10个cs以得到中间值的良好表示。此步骤通过以下等式有效进行:
xi,j=ri,j/中间值(ri,cs1:ri,csx)
其中ri,j是样品i在位点j处的原始读取数。中间值在cs位点的组中的所有位点j上进行评估。xi,j是在样品i在位点j处的“样品归一化深度值”;xi,j针对cs和ts两者中的所有位点j进行计算。
如图9中所提供的,表a中每个单元的值除以表b中单元的排的对应值,并且商记录在表c中。现在整个排的平均值是~1。然而,需要进一步归一化,因为在数据采集中存在可能破坏对数据的解读的位点特异性偏差。例如,注意tsx列中的值系统性地低于ts1或ts2中的值。因为在tsx处的此降低反映每个样品中的cn变化是不真实的(尤其是因为假设cn变异很少见,因此预期此类变异不在每个样品中),因此进行进一步归一化(在图9,表d中)来消除此系统性偏差。
归一化以计算每列下的中间值开始。如图9表d所示,这对于ts列和cs列两者均已完成。然后,如图9表e所示,表c中每个单元的值除以表d中每个单元的列的对应值;然后将商乘以二,并且最后将乘积记录在表e中。将商扩大2倍,因为除以平均值得到集中在1左右的归一化值,但是已知此归一化值对应于生物正常cn2。此步骤通过以下等式有效进行:
cni,j=2*xi,j/中间值(xs1,j:xsx,j)
其中xi,j是来自以上的“样品归一化深度值”。中间值是在所有样品上针对位点j进行计算。cni,j是样品i中位点j的拷贝数的小数近似值。因为基因组中序列的拷贝数是整数值,所以每个cni,j可四舍五入至其最接近的整数值,并且调用的置信度可如本文所述地进行计算。
应注意,在上述等式中所指示的最终归一化步骤可在cn高度可变的情况下(即,在微弱多数部分或甚至小部分样品具有cn=2的情况下)针对ts进行改变。例如,在图8的右曲线图中,大部分样品对于ts“wl5,b08”和“wl5,b09”而言具有cn=0而不是cn=2。已经在sma(图5)和cah(图8)的分析中遇到过此类ts。在这些有挑战性的ts处的cn值可通过找到多模态高斯分布(multimodalgaussiandistribution)(使用在凭经验预期的整数cn值例如0、1、2和3下的模式)与凭经验观察的数据的最佳最小平方偏差拟合来确定。每个样品的cn值然后可通过找到对最佳拟合分布的整数模式的最小距离来确定。
最后的步骤是对数据的解读。针对每种疾病(先天性肾上腺皮质增生症(cah)、脊髓性肌萎缩(sma)、戈谢氏病以及α地中海贫血),寻找cn信号偏离2的连续ts。应注意,与具有集中在2处的cn值的其他样品不同,图9中的“样品1”具有徘徊在1左右的cn值。这些数据表明样品1在ts处具有将其cn从二降到一的cn突变。令人安心的是看到样品1在cs处的cn值是~2,这表明分析是合理的(即,并不认为样品在基因组的任何位置处均具有cn突变,这是不真实的)。
值得注意的是,本文所述的cn分析是找到具有同源物的基因中其他类型的临床相关性突变的关键性上游步骤。例如,除cn变体(图1中所示)之外,单核苷酸多态性(snp)也可能破坏基因并且使其功能失调。用于识别snp的标准软件使用cn作为参数,其中具有snp的读取的预期分数是1/cn。因为基因组的大多数部分具有cn=2,所以当读取的1/2包含一个碱基(例如,c)并且另外1/2具有不同的碱基(例如,t)时,snp寻找软件通过默认方式将位点识别为snp。然而,对于具有cn变异的区域,具有snp的读取的预期分数可以是1(针对cn=1)、1/3(针对cn=3)等。关键地是,在cn分析如与本文所述的相同的分析不存在下,具有snp和cn=3两者的受试者可能不能使snp被识别,因为其在数据中的表示(即,1/3)小于自然预期的分数(即,1/2)。因此,本文中所描述的方式不仅对于在cn方面解析基因型是重要的,而且对于在找到其他突变如snp和短插入/缺失(“插入缺失”)方面解析基因型也是重要的。
因为通常对于给定的测试具有多个ts,所以可使用z得分来估计cn确定的置信度。以下是可使用的步骤:
a.针对每个ts列计算四位分差(“iqr”)。iqr是第75个与第25个百分位值之间的差值。假设正态分布统计,通过除以~1.33来将iqr转换为标准偏差(“sd”)。使用iqr作为中间步骤以找到sd,因为iqr对于离群值是不敏感的,而sd可随着离群值大幅度变化。这种对于离群值的关注是尤其重要的,因为具有cn突变的少见样品实际上是每列中的离群值。
b.在得到每个ts列的sd的情况下,接下来列举猜想(即,cn=1、cn=2等),并且针对每个猜想,确定观察到的cn值的sd远离猜想的cn值多少(sd距离假设的平均值的这个数是z得分)。接下来,可将z得分转换为概率,所述概率使得能够鉴于数据评估所述猜想的可能性。将每个位点作为独立的观察进行处理,将穿过许多ts的概率计算为每个ts的概率的乘积。置信度得分实际上是对数差别得分(log-oddsscore),其中将最高概率的猜想的概率除以第二高概率猜想的概率,并且然后取此商的对数10。
本领域的技术人员将认识到,可使用对离群值不敏感并产生数据的标准偏差的近似值的其他统计方式。对类似拷贝数的跨距(例如,具有cn=1的一系列相邻位点,与较大缺失一致)的识别可以监控的方式(例如,通过观看或通过匹配已知的或猜想的重组位点)或非监控的方式(例如,使用隐马尔可夫模型(hiddenmarkovmodel))来识别。
示例性架构和处理环境:
本文所述的系统和过程的某些方面和实施例可在其中运行的示例性环境和系统。如图3所示,在一些实施例中,所述系统可根据客户端-服务器模型(client-servermodel)来实现。所述系统可包括在用户装置102上执行的客户端部分和在服务器系统110上执行的服务器端部分。用户装置102可包括任何电子装置,诸如台式电脑、膝上型电脑、平板电脑、pda、移动手机(例如,智能手机)等。
用户装置102可通过一个或多个网络108与服务器系统110通信,所述网络108可包括因特网、内联网或任何其他有线或无线的公共网络或私人网络。用户装置102上的示例性系统的客户端部分可提供客户端功能性,诸如面向用户的输入和输出处理以及与服务器系统110的通信。服务器系统110可为驻留在相应的用户装置102上的任何数量的客户端提供服务器端功能性。另外,服务器系统110可包括一个或多个呼叫者服务器114,所述呼叫者服务器114可包括面向客户端的i/o界面122、一个或多个处理模块118、数据和模型储存器120以及外部服务的i/o界面116。面向客户端的i/o界面122可促进呼叫者服务器114的面向客户端的输入和输出处理。一个或多个处理模块118可包括如本文所述的各种问题和候选者得分模型。在一些实施例中,呼叫者服务器114可通过网络108与外部服务124(诸如文本数据库、订阅服务、政府记录服务等)通信以用于完成任务或采集信息。外部服务的i/o界面116可促进此类通信。
服务器系统110可在一个或多个独立的数据处理装置或计算机的分布式网络上实现。在一些实施例中,服务器系统110可采用第三方服务提供商(即,第三方云服务提供商)的各种虚拟装置和/或服务来提供服务器系统110的基础计算资源和/或基础设施资源。
虽然呼叫者服务器114的功能性在图3中示出为包括客户端部分和服务器端部分两者,但是在一些实施例中,本文所述的某些功能(例如,关于用户界面特征和图形元素)可作为安装在用户装置上的独立应用来实现。此外,功能性在系统的客户端部分和服务器部分之间的划分可在不同实施例中变化。例如,在一些实施例中,在用户装置102上执行的客户端可以是瘦客户端,所述瘦客户端仅提供面向用户的输入和输出处理功能,并且向后端服务器授权系统的所有其他功能性。
应注意,服务器系统110和客户端102还可包括各种类型的计算机装置中的任一种,其具有例如处理单元、存储器(其可包括用于实施本文所述的功能中的一些或所有的逻辑或软件)和通信界面以及其他常规计算机部件(例如,输入装置(诸如键盘/触摸屏)和输出装置(诸如显示器))。另外,服务器系统110和客户端102中的一者或两者通常包括逻辑(例如,http网络服务器逻辑),或者被编程为格式化数据,所述格式化数据从本地或远程数据库或其他数据和内容物来源访问。为此,服务器系统110可利用各种网络数据界面技术,诸如公共网关接口(cgi)协议和相关应用程序(或“脚本”)、
还应注意,虽然本文所述的示例性方法和系统描述了用于执行各种功能的单独的服务器和数据库系统的使用,但是其他实施方案可通过存储操作来在单个装置或多个装置的任何组合(作为设计选择,只要执行所描述的功能性即可)上引起所描述的功能的软件或编程来实现。类似地,所描述的数据库系统可实现为单个数据库、分布式数据库、分布式数据库的集合、具有冗余的在线或离线备份或其他冗余的数据库等,并且可包括分布式数据库或存储网络和相关处理智能。虽然没有在图中描绘出,但是服务器系统110(和本文所述的其他服务器和服务)通常包括如在服务器系统中常见的此类本领域认可的部件,包括但不限于处理器、ram、rom、计时器、硬件驱动器、相关储存器等(参见例如图4,在以下进行讨论)。另外,所描述的功能和逻辑可包括在软件、硬件、固件或其组合中。
图4描绘示例性计算系统600,所述计算系统600被配置来执行以上所述的过程中的任一个,包括各种调用和得分模型。在此情况下,计算系统600可包括例如处理器、存储器、储存器以及输入/输出装置(例如,监测器、键盘、磁盘驱动器、因特网连接件等)。然而,计算系统600可包括用于实施所述过程的一些或所有方面的电路或其他专用硬件。在一些操作性设置中,计算系统600可被配置为包括一个或多个单元的系统,每个所述单元被配置来在软件、硬件或其某一组合中实施所述过程的一些方面。
图4描绘具有可用于执行以上所述的过程的多个部件的计算系统600。主要系统1402包括母板1404,所述母板1404具有输入/输出(“i/o”)部分1406、一个或多个中央处理单元(“cpu”)1408以及存储器部分1410,所述存储器部分1410可具有与其相联的闪存卡1412。i/o部分1406连接到显示器1424、键盘1414、磁盘存储单元1416以及媒体驱动单元1418。媒体驱动单元1418可读取/编写计算机可读介质1420,所述计算机可读介质1420可包含程序1422和/或数据。
基于以上所述过程的结果的至少一些值可保存以供后续使用。另外,非暂时性计算机可读介质可用于存储(例如,有形地体现)用于通过计算机执行以上所述的过程中的任一个的一个或多个计算机程序。计算机程序可例如以通用编程语言(例如,pascal、c、c++、python、java)或某一专用的特定应用语言来编写。
本文描述了各种示例性实施方案。以非限制性意义对这些实施例进行参考。它们被提供来说明所公开的技术的可更广泛应用的方面。在不背离各种实施方案的正确精神和范围的情况下,可做出各种改变并且可取代等同物。此外,为了使特定的场合、材料、物质组成、方法、方法行为或步骤适应各种实施方案的目的、精神和范围,可进行许多修改。另外,如本领域技术人员将认识到的,本文所描述并说明的每个单个变型具有离散组分和特征,所述离散组分和特征可易于与任何其他多个实施方案的特征分离或组合,而不背离各种实施方案的范围或精神。所有此类修改意图处于与本公开相关联的权利要求书的范围内。
实施例
本发明在以下实施例中进一步详细描述,所述实施例不意图以任何方式限制要求保护的本发明范围。附图旨在被视为本发明的说明书和描述的组成部分。引用的所有参考文献针对其中描述的所有内容特定地以引用的方式并入本文。提供以下实施例来说明,但并非限制所要求保护的发明。
实施例1
调用基因/同源物拷贝数
此实施例说明了用于确定基因/同源物拷贝数的方法并且在图9中示意性地示出。
所述方法包括以下步骤。
1.合并bwa(将ngs读取与参考基因组比对的开放源码计算机软件程序)分配给基因或同源物的所有读取。
2.基于读取的序列(任选地调整读取深度以将gc偏差考虑在内),分别计数基因和同源物(例如,在辨别smn1与smn2的内含子位置处)的深度(即,比对读取的数)。
3.计算50个其他对照位点(图9中的“cs”)附近的深度
4.通过样品的50个对照深度的中间值对每个样品的基因和同源物深度进行归一化。
5.通过由每个位点的中间值进行归一化来进一步调整数据,从而产生基于小数的拷贝数值(例如,1.21)。
6.基于对置信度的统计学评估进行拷贝数调用(即,将来自前一步骤的小数值映射为整数值)。
各种基因/同源物确定的结果在图5-图8中示出。
实施例2
使用杂交捕获探针的拷贝数分析
此实施例说明了用于使用探针确定特定基因的基因/同源物拷贝数的方法,所述探针与在基因与同源物或假基因之间不同的碱基相邻退火。
杂交捕获探针被设计来与在cyp21a2与cyp21a1p之间不同的几个碱基(“差别碱基”)相邻地退火。所捕获的片段的末端配对的ngs允许基于差别碱基将读取指定为基因衍生的或假基因衍生的。使用以下两个策略来识别cah变体:基于snp的调用和拷贝数分析。给定位置处基于snp的调用在由具有远离感兴趣位置的基因衍生的差别碱基的读取构成的堆叠中查找有害的和/或假基因衍生的碱基。相比之下,拷贝数分析使用差别碱基的读取深度来计算每个变体的相对丰度,并且有害变体被识别为具有过多拷贝数(以及,相反地,缺失拷贝数的基因衍生的序列)的假基因衍生的序列的那些变体。使用长范围pcr和sanger测序来以验证研究确认变体。
测试正确地从受影响患者中识别阳性对照样品的基因型,并且已经对将近150,000个临床样品进行了验证的cah测试。所观察到的变体频率与测序受影响患者的cyp21a2的先前研究一致。在基因和假基因的拷贝数中存在极大的多样性:38%的患者具有至少一个单倍型,所述至少一个单倍型不仅具有基因和假基因的一个拷贝。基因与假基因之间的重组的证据是广泛的,其中至少83%具有包含假基因衍生的碱基的cyp21a2单倍型。最后,测试识别了与特定少见的单倍型一致的化合物变体,例如,(1)三个cyp21a2拷贝,其中一个具有q319x突变,以及(2)具有顺式v282l突变的cyp21a2和两个cyp21a1p拷贝,单倍型富集在德系犹太人(ashkenazijewish)患者中。
应理解,本文所述的实施例和实施方案仅出于说明性目的,并且将建议本领域技术人员根据它们进行各种修改或改变,并且它们被包括在本申请的精神和权限以及随附权利要求书的范围之内。出于所有目的,本文所引用的所有出版物、专利和专利申请特此以引用的方式整体并入。
- 还没有人留言评论。精彩留言会获得点赞!