软固体结晶电解质组合物的制作方法
本申请要求2014年3月25日提交的系列号为61/970,146的美国专利申请的优先权,所述美国专利申请的完整内容以引用的方式并入本文。
有关联邦政府发起的研究或开发的声明
本发明是在政府支持下根据由美国国家科学基金会(National Science Foundation)授权的授权号DMR 1207221和CBET-1437814进行。政府对本发明享有某些权利。
发明背景
目前用于如锂/锂离子电池、氢离子燃料电池和太阳能电池的电化学装置中的电解质典型地为液体或凝胶电解质。然而,尽管具有>1×10-3S/cm的良好室温电导率,这些液体或凝胶电解质具有安全考虑,如泄漏、归因于挥发性溶剂的爆炸、树枝状晶体形成以及比在固体电解质中快的降解产物的形成/迁移(Xu,K.,Nonaqueous,2004,Chemical Reviews 104,(10),4303-4417;Aurbach,D.;Zinigrad,E.;Cohen,Y.;Teller,H.,Solid State Ionics 2002,148,(3-4),405-416;Brissot,C.;Rosso,M.;Chazalviel,J.N.;Lascaud,S.,J of the Electrochemical Society 1999,146,(12),4393-4400)。因此,需要具有在宽泛温度范围内促进增强的离子迁移的结构的新材料来替换电化学装置中的这些可燃液体或凝胶电解质。
固态电解质先前已经由于与固态材料有关的安全性的预期增加而进行研究,但这些电解质典型地具有相对不良的离子电导率(Zaghib等人,2011,J of Power Sources 196,3949-3954)。目前可得的具有最高离子电导率的固体电解质为陶瓷/玻璃和其它无机超离子导体,其中电导率潜在地在10-3至10-2S/cm范围内(Fergus,2010,J of Power Sources 195,4554-4569)。在无机超离子导体的情况下,结晶系统典型地比玻璃更具导电性(Kanno和Maruyama,2001,Journal of the Electrochemical Society 148(7),A742-A746)。关于固体电解质最初报道的具有高RT离子电导率(6×10-3S/cm)的Li+离子超离子导体Li3N具有低电化学稳定性窗口,使得其不适合作为固体电解质(Alpen等人,1977,Applied Physics Letters 30(12),621-62;Lapp等人,1983,Solid State Ionics 11(2),97-103)。其它无机超离子固体电解质具有较佳的电化学稳定性,但具有较低的离子电导率(~10-3S/cm),如结晶氧化物钙钛矿型钛酸锂镧(La0.5Li0.5TiO3)(Inaguma等人,1993,Solid State Communications 86(10),689-693)、一系列硫化物晶体(如具有γ-Li3PO4的构架结构的Li4-xGe1-xPxS4,被称作硫代-LISICON(例如Li3.25Ge0.25P0.75S4))(Kanno和Maruyama,2001,Journal of the Electrochemical Society 148(7),A742-A746)、玻璃陶瓷(70Li2S-30P2S5)(Mizuno等人,2005,Advanced Materials 17(7),918-921;Hayashi等人,2008,Journal of Materials Science 43(6),1885-1889)和玻璃状材料(Li2S-SiS2-Li3PO4)(Kondo等人,1992 Solid State Ionics 53,1183-1186;Takada等人,1993,Journal of Power Sources 43(1-3),135-141)。仅Li2.9PO3.3N0.46(LiPON)在市面上用作微电池组中的固体电解质(Bates等人,1992,Solid State Ionics 53,647-654;Bates等人,1993,Journal of Power Sources 43(1-3),103-110)。关于锂超离子导体的最高RT离子电导率近来已经关于Li10GeP2S12(12mS/cm)进行报道。用Sn取代Ge也形成超离子晶体Li10SnP2S12(7mS/cm),并且两种材料均为亚稳的(Bron等人,2013,J Am Chem Soc 135(42),15694-15697;Mo等人,2012,Chemistry of Materials 24(1),15-17)。然而,这些电解质为脆性的,并且其由于在连续充电/放电循环期间的体积改变而对电极具有不良粘着。
软固体电解质展现所需的柔性,但具有低于陶瓷/玻璃/无机导体的电导率(例如,在10-7至10-5S/cm范围内的电导率)。软固体电解质的实例包括聚环氧乙烷(PEO)(Abitelli等人,2010,Electrochimica Acta 55,5478-5484)、PEO/复合共混物(Croce等人,1998,Nature 394,456-458;Croce等人,1999,J of Physical Chemistry B 103,10632-10638;Stephan等人,2009,J of Physical Chemistry B 113,1963-1971;Zhang等人,2010,Electrochimica Acta 55,5966-5974;Zhang等人,2011,Materials Chemistry and Physics 121,511-518;Zhan等人,2011,J of Applied Electrochemistry 40,1475-1481;Uvarov,2011,J of Solid State Electrochemistry 15,367-389)、PEO共聚物/共混物(Tsuchida等人,1988,Macromolecules 21,96-100;Ryu等人,2005,J of the Electrochemical Society 152,A158-A163;Park等人,2004,Electrochimica Acta 50,375-378)、分子或离子塑性晶体(Timmermans,1961,J of Physics and Chemistry of Solids 18,1-8;Sherwood,1979,The Plastically Crystalline State:Orientationally Disordered Crystals,Wiley,Chichester,UK;MacFarlane和Forsyth,2001,Advanced Materials 13,957-966;Pringle等人,2010,J of Materials Chemistry 20,2056-2062;Cooper和Angell,1986,Solid State Ionics 18-9,570-576;Yoshizawa-Fujita等人,2007,Electrochemistry Communications 9,1202-1205.)和低分子量甘醇二甲醚(Henderson等人,2003,Chemistry of Materials 15,4679-4684;Henderson等人,2003,Chemistry of Materials 15,4685-4690;Seneyiratne等人,2004,J of Physical Chemistry B 108,8124-8128;Andreev等人,2005,Chemistry of Materials 17,767-772;Henderson等人,2005,Chemistry of Materials 17,2284-2289;Henderson,2006,J of Physical Chemistry B 110,13177-13183;Zhang等人,2007,Angewandte Chemie-International Edition 46,2848-2850;Zhang等人,2007,J of the American Chemical Society 129,8700-8701)。软固体电解质材料的另一实例为NAFIONTM聚合物,其具有含有阴离子涂布(典型地-SO3-)的渗透簇的疏水性全氟化基质,和带相反电荷的离子可迁移通过的通道(Mauritz和Moore,2004,Chemical Reviews 104,4535-4586)。
关于PEO系统,导电性已经显示主要通过非晶相出现,其中结合离子迁移来减慢骨架区段运动(Borodin和Smith,2006,Macromolecules 39,1620-1629),使得结晶度的降低(Abitelli等人,2010,Electrochimica Acta 55,5478-5484;Stephan等人,2009,J of Physical Chemistry B 113,1963-1971;Zhang等人,2010,Electrochimica Acta 55,5966-5974;Zhan等人,2011,J of Applied Electrochemistry 40,1475-1481)和聚合物链的对准(Bruce,1996,Philosophical Transactions of the Royal Society a-Mathematical Physical and Engineering Sciences 354,1577-1593;Andreev和Bruce,2000,Electrochimica Acta 45,1417-1423)会增加电导率。
改进软固体电解质中的离子导电性的其它方法是基于以下观察:分子组织化而非无序结构促进离子迁移率。具体说来,这对于其中存在用于离子迁移的替代、低活化能路径的材料确实如此,所述路径如沿着并且在组织化、对准的聚合物或液晶聚合物链之间(Andreev和Bruce,2000,Electrochimica Acta 45,1417-1423;Golodnitsky和Peled,2000,Electrochimica Acta 45,1431-1436;Dias等人,1998,Electrochimica Acta 43,1217-1224;Hubbard等人,1998,Electrochimica Acta 43,1239-1245;Imrie等人,1999,Advanced Materials 11,832-834;Yoshio等人,2004,J of the American Chemical Society 126,994-995;Kishimoto等人,2003,J of the American Chemical Society 125,3196-3197;Yoshio,2006,J of the American Chemical Society 128,5570-5577;Shimura等人,2008,J of the American Chemical Society 130,1759-1765;Ichikawa,2011,J of the American Chemical Society 133,2163-2169);沿着聚合物/无机纳米粒子界面,可能归因于醚O-Li+键的弱化(Shen,2009,Electrochimica Acta 54,3490-3494;Chen-Yang等人,2008,J of Power Sources 182,340-348;Marcinek等人,2000,J of Physical Chemistry B 104,11088-11093;Borodin等人,2003,Macromolecules 36,7873-7883);和沿着低分子量甘醇二甲醚和三锂化合物中的离子通道(Gadjourova等人,2001,Nature 412,520-523;MacGlashan等人,1999,Nature 398,792-794;Gadjourova等人,2001,Chemistry of Materials 13,1282-1285;Stoeva等人,2003,J of the American Chemical Society 125,4619-4626;Staunton等人,2005,J of the American Chemical Society 127,12176-12177;Zhang等人,2007,J of the American Chemical Society 129,8700-8701;Zhang等人,2008,Chemistry of Materials 20,4039-4044;Moriya等人,2012,Chemistry-A European J 18,15305-15309)。在可移动阳离子(如Li+)与其缔合的阴离子和/或溶剂化基质之间的降低的相互作用(如在微相分离的固体聚合物电解质(SPE)中)也已经显示增加阳离子迁移率和电导率(Ryu等人,2005,J of the Electrochemical Society 152,A158-A163)。关于具有较高电导率的软固体电解质的设计,需要其中通道壁对封闭的离子具有低亲和力的结晶固体。
除了当用于空气或液体流通阴极中时对于稳定性窗口和与溶剂的相容性的一般考虑以外,关于固体电解质在所有固态Li电池中的使用保留的关键问题为室温离子电导率的改进、增加的充电/放电速率、用于避免极化效应的高锂离子迁移数和在重复的充电/放电循环期间在电极中出现的体积改变期间良好电极/电解质接触的维持(Doyle等人,1994,Electrochimica Acta 39,(13),2073-81;Thomas等人,2000,J of Power Sources 89,(2),132-138;Gadjourova等人,2001,Nature 412,(6846),520-523)。具有可增强离子迁移的具体离子导电路径的固态有机材料的工程改造提供作为实现较高固态离子电导率的方式的希望,而软的、更具延展性的有机物可更佳地粘附于电极。然而,在这一领域中仅存在有限的进展。
另外,关于钠离子电池的材料研究已经集中于阴极,所述研究是极其需要的,因为钠比锂更丰富(Lu等人,2010,J of Power Sources 195,(9),2431-2442;Lu等人,2010,JOM 62,(9),31-36;Kim等人,2012,Advanced Energy Materials 2,(7),710-721;Ellis和Nazar,2012,Current Opinion in Solid State & Materials Science 16,(4),168-177;Palomares等人,2012,Energy & Environmental Science 2012,5,(3),5884-5901)。关于用于钠离子电池的电解质存在较少研究,对于所述电解质来说,钠盐在质子惰性溶剂中的可溶性低于锂盐。如NASICON Na+超离子导体的无机材料为所研究的最常见固体钠电解质(Fuentes等人,2001,Solid State Ionics 140,(1-2),173-179)。然而,关于聚环氧乙烷(Ma等人,1993,J of the Electrochemical Society 140,(10),2726-2733;Mohan等人,2007,Soft Materials 5,(1),33-46;Mohan等人,2007,J of Polymer Research 14,(4),283-290;Kumar等人,2011,Physica B-Condensed Matter 406,(9),1706-1712)或基于聚乙烯醇的聚合物电解质(Bhargav等人,2007,Ionics 13,(3),173-178;Bhargav等人,2007,Ionics 2007,13,(6),441-446)以及聚合物凝胶(Kumar和Hashmi,S.A.,2010,Solid State Ionics 181,(8-10),416-423;Kumar等人,2011,Solid State Ionics 202,(1),45-53;Kumar等人,2011,Solid State Ionics 201,(1),73-80)也存在研究。关于Na-离子固体电解质的相同考虑和需求对于Li-离子固体电解质来说是必需的。
因此,本领域中持续需要用于电化学装置的固态电解质,包括固体钠电解质。本发明解决了本领域中的这一持续需要。
发明概述
本发明涉及软固体电解质组合物和用于制造所述组合物的方法。在一个实施方案中,本发明的组合物为包含离子化合物和有机化合物的共晶体的软固体电解质组合物。在一个实施方案中,所述共晶体包含离子通道。
在一个实施方案中,本发明的组合物为自支撑薄膜电解质组合物,其包含本发明的软固体电解质组合物的任一实施方案,和粘合剂。在一个实施方案中,所述粘合剂为聚环氧乙烷(PEO)。在另一实施方案中,所述粘合剂为用八条聚乙二醇链官能化的聚八面体倍半硅氧烷(POSS-PEG8)。
在一个实施方案中,本发明为一种用于制备软固体电解质组合物的方法,所述方法包括以下步骤:将离子化合物溶解于有机化合物中以形成溶液,和添加沉淀剂至所述溶液中,其中所述离子化合物和所述有机化合物的共晶体从所述溶液沉淀。在另一实施方案中,本发明的方法为一种用于制备软固体电解质组合物的方法,所述方法包括以下步骤:将离子化合物溶解于有机化合物中以形成溶液,和降低所述溶液的温度,其中在冷却时所述离子化合物和所述有机化合物的共晶体从所述溶液沉淀。在另一实施方案中,所述方法为用于制备软固体电解质组合物的方法,所述方法包括以下步骤:混合离子化合物与有机化合物以形成混合物,加热所述混合物以形成所述离子化合物于所述有机化合物中的溶液;和冷却所述溶液,其中在冷却时所述离子化合物和所述有机化合物的共晶体从所述溶液沉淀。
在一个实施方案中,本发明的方法可进一步包括分离所述共晶体的步骤。在另一实施方案中,所述方法可进一步包括保护所述共晶体免于空气影响的步骤。在另一实施方案中,所述方法可进一步包括保护所述共晶体免于水影响的步骤。在一个实施方案中,所述有机化合物是可聚合的。在一个实施方案中,所述方法进一步包括聚合共晶体的至少一部分的步骤。在一个实施方案中,所述沉淀剂为二乙醚(Et2O)。在多个实施方案中,由本发明的方法制造的共晶体包含离子通道。
在一个实施方案中,本发明的方法为一种用于制备自支撑薄膜电解质组合物的方法,所述方法包括以下步骤:混合粘合剂与溶剂以形成溶液,添加本发明的软固体电解质组合物的一个实施方案至所述溶液中,和去除所述溶剂以形成自支撑薄膜。在一个实施方案中,所述溶剂为二乙醚。在一个实施方案中,所述粘合剂为PEO或POSS-PEG8。
在一个实施方案中,本发明的组合物具有至少约1×10-5S/cm的电导率。在多个实施方案中,本发明的离子化合物为锂盐。在一个实施方案中,所述锂盐为氯化锂。在多个实施方案中,所述离子化合物为钠盐。在一个实施方案中,所述钠盐为高氯酸钠。在另一实施方案中,所述钠盐为六氟磷酸钠。
在多个实施方案中,本发明的有机化合物为软路易斯供体。在一个实施方案中,所述有机化合物为有机溶剂。在一个实施方案中,所述有机溶剂为N,N-二甲基甲酰胺(DMF)。在另一实施方案中,所述有机溶剂为吡啶。在一些实施方案中,所述有机化合物为羰基化合物。在一个实施方案中,所述羰基化合物选自由二苯甲酮、苯乙酮、苯甲酸苯酯组成的组。在另一实施方案中,所述有机化合物是选自由吡啶和异喹啉组成的组的吡啶基衍生物。在一些实施方案中,所述有机化合物为芳族烃。在一个实施方案中,所述芳族烃选自由二苯基甲烷(DPM)、三苯基甲烷、联苄、联苯和萘组成的组。
附图简述
本发明的优选实施方案的以下详述在结合附图阅读时将更好地得到理解。出于说明本发明的目的,在附图中显示目前优选的实施方案。然而,应理解,本发明不限于附图中所示的实施方案的精确安排和手段。
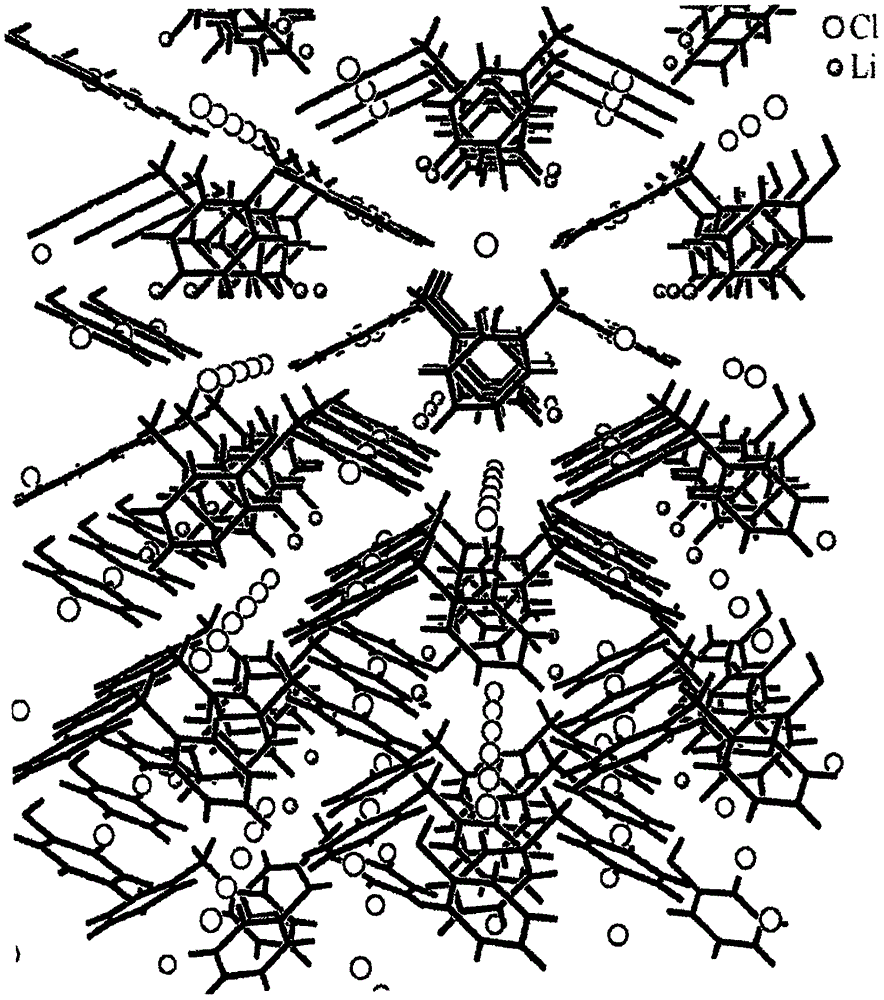
图1包括图1A和1B,是沿着a轴检视的N,N-二甲基甲酰胺(DMF):LiCl(图1A)和吡啶(py):LiCl(图1B)的晶体堆积图的集合。Li+和Cl-离子的通道在两个结构中均为明显的。
图2为二苯甲基(DPM):LiCl共晶体的晶体堆积的图,示出与二苯基甲烷的C-H键仅具有微弱相互作用的平行Li+和Cl-离子通道。
图3包括图3A至3F,是包含Na+盐的本发明组合物的示例性实施方案的晶体堆积图和X射线晶体结构的集合。
图4为说明本发明的离子-基质组装方法的图。
图5为通过烯烃官能团的紫外线诱导的自由基聚合使4-乙烯基吡啶和LiCl的软固体共结晶基质聚合的反应方案的说明。
图6包括图6A和6B,是DMF:LiCl(图6A)和Py:LiCl(图6B)的热椭球曲线的集合,说明Li2Cl2菱面体与对称等效的相邻菱面体和溶剂分子的相互作用。椭球被设定于50%概率水平下并且氢原子显示为空心圆。
图7为DMF(底部曲线)、溶解于LiCl中的DMF(中间曲线)和DMF:LiCl复合物(顶部曲线)的AT-IR光谱,示出在LiCl存在下在1690cm-1附近C=O波段的红移。
图8为与PEO/LiX;Li(BETI)=LiN(SO2CF2CF3)2;Li(TFSI)=LiN(SO2CF3)2相比LiCl·DMF的电导率对温度曲线(关于曲线中包括的文献值数据,参看Fergus,2010,J.of Power Sources,195:4554.;Jannasch,2002,P.Chemistry of Materials,14:2718)。
图9为DMF:LiCl的经过标记的粉末谱图的集合。顶部曲线:源于DMF:LiCl的单晶X射线结构的理论谱图。从顶部起的第二曲线:在电导率测量之前的大块样品。从顶部起的第三曲线:在电导率测量之后的大块样品。底部曲线:具有10%POSS-PEG8(用八条聚乙二醇链官能化的聚八面体倍半硅氧烷(POSS))的粉末样品。
图10为py:LiCl的粉末谱图的集合。底部曲线:源于py:LiCl的单晶X射线结构的理论谱图。顶部曲线:大块样品。
图11为示出DMF:LiCl(右侧峰)和Py:LiCl(左侧峰)的热重分析(TGA)曲线的图。
图12为LiCl晶体加合物的TGA曲线。
图13为具有相邻对称等效物的py·LiCl的热椭球曲线。椭球被设定于50%概率水平下。氢显示为空心圆。
图14为具有相邻对称等效物的DMF·LiCl的热椭球曲线。椭球被设定于50%概率水平下。氢显示为空心圆。
图15为用八条聚乙二醇链官能化的聚八面体倍半硅氧烷(POSS-PEG8)的结构图。
图16为顶部:具有10%POSS-PEG8的DMF-LiCl和底部:具有60%POSS-PEG8的DMF-LiCl的差示扫描热量测定曲线。
图17为针对经过压制的具有10%POSS-PEG的DMF-LiCl和Py-LiCl球粒的加热和冷却的温度依赖性电导率曲线。
图18为与LiCl:DMF溶液电解质相比并且与所示POSS-PEG8的10%POSS-PEG8结构相比的LiCl·DMF的电导率对温度曲线。
图19为用于通过使用阿伦尼乌斯方程来测量DMF:LiCl的活化能的log电导率(σ)对1/T图的曲线。
图20包括图20A至20C,是针对以下的曲线的集合:A)具有10%POSS-PEG8/Li°的Li°/DMF:LiCl在15℃.下在DC极化电压30mv下随时间而变的DC电流;B)最初和在稳态电流之后取得的科尔-科尔曲线,和C)具有10%POSS-PEG8/Li°的Li°/DMF:LiCl在15℃下在开路条件下随存储时间而变的所选科尔-科尔曲线。
详述
应了解,本发明的图式和描述已经简化以说明与本发明的清楚理解有关的要素,同时出于清晰目的消除在本领域中发现的与有机-离子组合物、电池组技术、适用于电池或其它电化学装置的电解质等有关的多种其它要素。所属领域的技术人员可认识到其它要素和/或步骤是实施本发明可需要的和/或需要的。然而,因为所述要素和步骤是本领域中众所周知的,并且因为其不会促进本发明的更佳理解,所以本文中未提供所述要素和步骤的论述。本文公开内容是针对所属领域的技术人员已知对所述要素和方法进行的所有所述变化和修改。
除非另外定义,否则本文所使用的所有技术和科学术语都具有与本发明所属领域的技术人员通常所理解相同的含义。尽管类似于或等效于本文所述的那些的任何方法、材料和组分可用于实践或测试本发明,但描述优选方法和材料。
如本文所用,以下术语中的每一者均具有在本章节中与其有关的含义。
冠词“一(a)”和“一(an)”在本文中用于指一个或超过一个(即至少一个)所述冠词的语法对象。举例来说,“一要素”意指一个要素或超过一个要素。
如本文所用的“约”当提及如量、时距等的可测量值时意图涵盖距规定值±20%、±10%、±5%、±1%或±0.1%的变化,因为所述变化是适当的。
在本公开中,本发明的各个方面可以范围形式呈递。应理解,呈范围形式的描述仅出于便利并且简洁目的并且不应被解释为对本发明的范围的坚定限制。相应地,范围的描述应被视为具有具体公开的所有可能的子范围以及在所述范围内的个别数值。例如,如1至6的范围的描述应被视为具有具体公开的子范围,如1至3、1至4、1至5、2至4、2至6、3至6等,以及在所述范围内的个别数字,例如1、2、2.7、3、4、5、5.3、6和其间的任何全增量和偏增量。与所述范围的宽度无关,这均适用。
描述
本发明涉及包含由有机基质围绕的离子化合物的组合物,和用于制造所述组合物的方法。所述组合物含有独特地低亲和力离子通道,并且在室温下一般为固体。在一个实施方案中,所述组合物为包含如锂或钠的离子的软固体电解质,其可用于电池。在另一实施方案中,所述组合物可包含除锂或钠外的离子,并且可用于非电池技术,如水纯化、燃料电池和催化装置。值得注意地,本发明的组合物可使用多种行业通常使用的廉价有机溶剂制备。本文描述具有离子通道的离子化合物和有机化合物的共晶体的成功形成的首次证实。
本发明的组合物和方法是基于以下理解:结晶固体电解质中的离子迁移率一般受到所述盐与周围基质的强离子相互作用阻碍,即固体中的离子迁移与高焓有关。然而,固体电解质中的离子迁移率可通过制造在所述盐与所述周围基质之间具有较弱偶极或甚至非极性相互作用的组合物来增强。因此,具有在所述盐与周围有机相之间具有弱相互作用的离子通道的有机/盐晶体可为需要的。相应地,本发明的组合物由基于皮尔森硬-软酸碱概念(HSAB)作为软路易斯供体并且与硬离子相对不良地相互作用的有机化合物制得。相应地,本发明的组合物一般为用于离子导电的结晶、软固体电解质,其中所述离子对周围基质具有低亲和力。
在多个实施方案中,本发明的组合物为有机化合物与盐的共晶体。所述有机化合物形成具有通道结构的基质,其中所述有机基质相对不良地与所述盐相互作用,因此允许通过所述通道结构的优秀离子迁移率。所得组合物具有相对高的离子电导率,例如大于10-5S/cm的电导率。进一步地,本发明的组合物的电导率在极宽范围内接近温度独立,指示关于离子迁移可忽略的活化障碍,即所述组合物展现无障碍离子导电。另外,本发明的电解质组合物在极低温度下(例如在-210℃或更低下)展现良好电导率,使得其适用于多种低温应用。
组合物
本发明涉及包含盐(即离子化合物)和有机化合物的共晶体的组合物。所述晶体一般为软固体,并且具有含于由所述有机化合物形成的基质内的所述盐的离子通道。
本发明的组合物是基于以下理解:当有机化合物使用本发明的方法与离子化合物共结晶时,源于离子-基质和离子-离子吸引力的额外晶格能组分导致形成具有无机电解质基质的高结晶度但具有聚合物电解质基质的柔软度和柔性的软固体。
在一个实施方案中,本发明的组合物的离子化合物可为锂或钠盐(即LiX或NaX,其中X=阴离子)。在多个实施方案中,所述盐的阴离子可为如所属领域的技术人员将了解的任何种类,包括但不限于:氯离子、溴离子、碘离子、四氟硼酸根、六氟磷酸根、高氯酸根、四苯基硼酸根、四(五氟苯基)硼酸根、[B[3,5-(CF3)2C6H3]4或[BArF4]-(“barf”)阴离子、在有机羧酸根(例如乙酸根、苯甲酸根)和有机磺酸根(例如PhSO3-)的类别中的阴离子。在另一实施方案中,所述盐可为除锂或钠盐外容易使用本发明的方法形成共晶体的任何类型的盐,例如但不限于:钾盐、镁盐或钙盐。在另一实施方案中,所述盐可包含超过一种类型的盐,即所述组合物可包含不同盐的混合物。
在多个实施方案中,本发明的组合物包含可与本文所述的盐形成共晶体的有机化合物。在多个实施方案中,适用于制备本发明的组合物的有机化合物为相对不良地与如Li+的硬离子相互作用的软路易斯供体。在一个实施方案中,所述有机化合物为有机溶剂,例如N,N-二甲基甲酰胺(DMF)或吡啶(Py)。所述有机溶剂典型地在室温下为液体。然而,本发明的组合物可包含一种或多种一般不被称作溶剂但作为软路易斯供体的有机化合物,如芳烃(例如二苯基甲烷(DPM));硫醇/硫化物(例如苯硫基甲烷);羰基化合物,包括酮、酯、酰胺和醛(例如二苯甲酮);腈(例如苯甲腈);膦(例如三苯基膦);或其路易斯酸供体能力通过HSAB概念描述为“软”的任何其它物质。这可包括在室温下并非液体的物质和/或化合物。适用于本发明的组合物的有机溶剂或化合物的其它实例包括但不限于:羰基化合物,例如二苯甲酮、苯乙酮、苯甲酸苯酯和吡啶基衍生物(例如吡啶和异喹啉);和芳族烃,例如三苯基甲烷、联苄、联苯和萘。然而,所述组合物可包含如所属领域的技术人员将了解的未在本文中具体列出的任何其它有机溶剂或化合物,包括本文列出的有机化合物的任何衍生物,例如甲基化衍生物。
此外,本发明的组合物包含关于离子导电最佳的结构。具体说来,所述组合物展现具有适用于离子导电的通道的有机基质的结构。在一个实施方案中,本发明涉及形成离子通道(其中LiX或NaX存在于有机通道中)的LiX-和NaX-有机晶体的形成,并且其中基质壁与LiX或NaX具有微弱极性、“软”(HSAB)或优选地非极性相互作用。在一个实施方案中,所述离子通道可为1-和2-D离子通道。在一个实施方案中,所述晶体可含有较高维度通道,即3-D通道或在2-D平面中的通道(在所述平面之间具有路径),因为1-D晶体中的点缺陷可限制离子迁移。
本发明的组合物的示例性实施方案包括氯化锂与DMF(DMF-LiCl)、吡啶(Py-LiCl)和二苯基甲烷(DPM-LiCl)的共晶体。这三个示例性实施方案的晶体堆积图在图1A(DMF-LiCl)、图1B(Py-LiCl)和图2(DPM-LiCl)中示出。进一步地,关于钠盐的共晶体的示例性实施方案的晶体堆积图在图3A((DMF)3-高氯酸钠,即每个NaClO4盐三个DMF分子)和图3B(Py4-六氟磷酸钠,即每个NaPF6盐四个吡啶分子)中示出。当沿着图1-3中的晶胞的a轴检视时,离子的线性通道是明显的。图3C和3D示出呈DMF中的固体溶液的NaClO4盐,其中钠离子通道垂直于页面(3C)并且单一通道平行于页面(3D)。图3E和3F示出呈吡啶中的固体溶液的NaPF6盐,其中钠离子通道垂直于页面(3E)并且单一通道平行于页面(3F)
本发明的组合物的特性和结构可通过有机化合物的选择来优化。一方面,所述有机化合物上的软路易斯供体的数目和类型可影响所得共晶体的特性和结构。另一方面,所得共晶体的特性和结构可依赖于所述有机化合物的其它特征,如所述有机化合物具有π-堆叠相互作用的能力。配体的细微差异先前已经显示出影响软晶体的晶体结构和传输特性(Zhang等人,2007,J of the American Chemical Society 129,(28),8700-8701)。因此,应理解所属领域的技术人员可选择和/或修改本发明的有机化合物以优化所述组合物的特性,如所述组合物的导电性、刚性和热稳定性。
例如,如在吡啶和二苯基甲烷LiCl复合物的基质中π-堆叠相互作用的增强所提示,芳基的使用可增强基质稳定性。在一个实施方案中,所述有机化合物可组合甲基化酰胺的溶剂化能力(即,如在DMF-LiCl加合物中所观察到)与在所述基质内形成较强π-堆叠相互作用的能力。所述酰胺的软羰基供体还提供与硬锂离子的微弱接触以允许轻易离子迁移。在所述实施方案中,所述有机化合物可为市面上有售的N,N-二甲基苯甲酰胺,或其类似物或衍生物。
同样,低熔点二苯甲酮、苯乙酮和苯甲酸苯酯的使用代表市面上有售的低熔点溶剂化物,其包括芳烃中π-堆叠相互作用的强度。关于基于吡啶的基质分子,异喹啉代表吡啶的低熔点固体类似物,其允许增加的π-堆叠机会。另外,与二苯基甲烷有关的烃(如三苯基甲烷、联苄、反芪、萘和联苯或其类似物或衍生物)可用于最小化离子-基质亲和力并且从π-堆叠增强基质稳定性。
在一个实施方案中,本发明的组合物的有机化合物可含有可聚合的乙烯基或其它烯系基团。在所述实施方案中,本发明的软-固体电解质的强度和稳定性可通过在有机基质化合物与所述离子化合物共结晶之后使所述有机基质化合物聚合来改进。例如,芳族烃苯乙烯或其衍生物可用于制备具有LiCl的共晶体组合物,并且接着所述共晶体组合物可经由光子活化和/或使用自由基或阴离子引发剂聚合。
本发明的组合物的特性和结构也可通过离子化合物的选择来优化。一方面,阳离子电导率可通过阴离子的变化,例如通过基于大小选择阴离子来增强。阴离子一般大于阳离子,并且其变化预期改变离子-基质共晶体中的结晶结构和通道大小至大于阳离子变化的程度。宽变化已经在具有多种平衡阴离子的Li甘醇二甲醚的结构和结晶动力学中观察到。这些盐的结晶动力学与质子惰性溶剂中不同锂盐的离子缔合行为良好相关(Henderson,2007,Macromolecules 40,(14),4963-4971)。一般来说,较大阴离子将归因于位阻不太自由地移动通过所述基质。然而,在一些情况下,较大阴离子可更自由地移动,因为其具有更具扩散性的电荷并且可形成大于较小阴离子的通道。因此,应理解所属领域的技术人员可视多种考虑因素选择离子化合物的平衡阴离子以优化或修改本发明的组合物的特性,所述考虑因素如所用的有机化合物或所述电解质的所需操作温度范围。
在一个实施方案中,所述组合物可包含两种或更多种离子化合物,即不同离子化合物的混合物。例如,具有相同阳离子(例如Li)但具有不同平衡阴离子的离子化合物可用于所述组合物。同样,在一个实施方案中,所述组合物可包含两种或更多种有机化合物,即不同有机化合物的混合物。因此,本发明的组合物的特性可通过在所述实施方案中选择不同离子或有机化合物的类型和/或相对量来进行修改或优化。
在一些实施方案中,本发明的组合物可展现离子导电和电子导电。例如,芳环的堆叠可产生用于离子导电的通道,而π电子云的重叠提供用于电子导电的路径。所述双重导电行为类似于电荷转移盐Li0.6(15-冠-5-醚)[Ni(dmit)2]2·H2O,所述盐展现经由镍复合物(dmit为有机分子)的堆叠产生的电子导电性,和经由提供用于锂离子移动的通道的所述冠醚的堆叠产生的离子导电性(Nakamura等人,1998,Nature 394(6689),159-162)。
在多个实施方案中,本发明的组合物可包含经由并入低重量百分比(例如10%)的离子导电材料产生的自支撑薄膜。例如,所述离子导电材料可为聚环氧乙烷(PEO)或用八条聚乙二醇链官能化的寡聚八面体倍半硅氧烷(POSS)(POSS-PEG8)。
方法
本发明的组合物可如下文所述合成。这些组合物的建构通过散布有有机基质分子的盐的简单晶体生长进行。本发明的方法可使用适合用作如LiX和NaX的盐的电解质基质的通常可得的有机化合物执行。所得组合物一般为具有优秀离子导电的软、柔性固体材料。
参看图4,示出用于制备本发明的组合物的方法的示例性实施方案的示意图。首先,在图4A中,如LiCl的离子化合物溶解于最少量的有机化合物(即溶剂)中,所述有机化合物例如DMF或吡啶。在极性(或可极化)溶剂存在下,离子化合物归因于用离子-偶极相互作用置换离子键而溶解,产生其中离子中散布有所述基质分子的溶液。其次,在图4B中,例如二乙醚(Et2O)的沉淀剂(precipitant/precipitating agent)添加至离子和基质分子的溶液中。在缓慢引入所述沉淀剂时,所述极性离子-分子组合件安排成组织化晶格以避免极性相与非极性相之间的分子相互作用(图4C),导致所述离子化合物和所述溶剂/基质分子的极纯共晶体的沉淀。结果为具有组织化并且充分分离的离子和高概率的离子通道的离子-分子基质。相对纯共晶体可接着与所述沉淀剂和/或任何未反应的起始材料分离。
相应地,在一个实施方案中,本发明的方法包括将离子化合物溶解于有机化合物中以形成溶液,和接着添加沉淀剂至所述溶液中的步骤,其中形成包含所述离子化合物和有机化合物的共晶体。在所述实施方案中,所述方法可进一步包括去除所述沉淀剂的步骤,例如如果所述沉淀剂为充分挥发性的,那么通过向所述组合物施加真空。在多个实施方案中,所述沉淀剂可为如所属领域的技术人员将了解的任何化合物,并且一般为可与本发明的有机化合物混溶的相对挥发性非极性溶剂。
本发明的组合物也可不使用沉淀剂合成。在一个实施方案中,所述离子化合物可溶解于所述有机化合物中,接着可通过降低所述溶液的温度形成所述离子化合物和有机化合物的共晶体。在另一实施方案中,所述离子化合物和有机化合物的共晶体可自发形成,而不需要降低所述溶液的温度。
在多个实施方案中,所述有机化合物可需要加热以促进所述离子化合物的溶解,或熔融所述有机化合物,例如当所述有机化合物在室温下为固体时。因此,本发明的方法可包括加热所述有机化合物、所述离子化合物或所述有机化合物和离子化合物的混合物的步骤。
由本发明的方法制造的晶体可归因于强路易斯酸性离子(例如Li+)极具吸湿性。相应地,本发明的结晶组合物当暴露于空气时可快速地吸收水以形成两相液体。然而,当所述晶体在相对冷温度下被存储于相对气密容器中时,所述晶体无限期地稳定。因此,本发明的方法可进一步包括保护所述晶体免于空气或水影响的步骤,例如通过转移共晶体至干燥箱或气密容器,和/或在合适温度下存储所述晶体,以便避免所述晶体的降解。
另外,使用本文所述的方法合成的共晶体可在形成后进行处理或修改。在一个实施方案中,所述共晶体中包含所述有机化合物的部分可聚合以改进所述组合物的稳定性或其它特征。相应地,本发明的方法可进一步包括聚合所述组合物的至少一部分的步骤,例如通过使共晶体暴露于紫外光和/或引发剂。本发明的有机化合物的聚合的实施例在图5中示出。在这一实施例中,4-乙烯基吡啶和LiCl的软固体共结晶基质经由紫外辐射聚合。
本发明进一步包括一种用于制备呈自支撑薄膜的本发明的共晶体的方法。在这种方法中,所述晶体被并入如PEO或POSS-PEG8的离子导电粘合剂组合物中。在一个实施方案中,所述方法包括以下步骤:将所述粘合剂(例如PEO或POSS-PEG8)溶解于溶剂中,其中所述溶剂不适合于溶解所述共晶体组合物;混合所述共晶体组合物与粘合剂/溶剂溶液;接着蒸发所述溶剂以形成包含所述粘合剂和共晶体组合物的薄膜。
实验实施例
本发明进一步通过参考以下实验实施例详细地描述。除非另外规定,否则这些实施例仅提供用于说明目的,并且不意图具限制性。因此,本发明绝不应视为限于以下实施例,而是应视为涵盖由于本文提供的教示而变得明显的任何和所有变化。
无需进一步描述,相信所属领域的技术人员可使用先前描述和以下说明性实施例制备并且利用本发明的组合物并且实践所要求的方法。以下工作实施例因此具体地指出本发明的优选实施方案,并且不应视为以任何方式限制本公开的剩余部分。
实施例1:DMF-LiCl和Py-LiCl共晶体
由常见、廉价实验室溶剂制备结晶软固体电解质。Li+和Cl-于结晶有机基质中的固体溶液的制备通过使共晶体从LiCl溶液沉淀来实现。LiCl(s)溶解于最少的溶剂(DMF或吡啶)中。二乙醚沉淀剂的添加导致LiCl和相应溶剂的纯白色共晶体的沉淀。这种溶剂组合对于纯材料的分离是理想的,因为溶剂DMF和吡啶均可溶(可混溶)于Et2O中,而LiCl不溶于Et2O中。据推测,Li+和Cl-离子呈溶剂加合物存在于溶液中,并且因此,当添加Et2O时,LiCl未呈纯盐而是呈与其溶剂的加合物沉淀,从而产生结晶、固体溶剂基质中的固体Li+和Cl-溶液
所得有机-离子共结晶晶格为固体材料,其具有相当良好的固态离子电导率,尤其在低温下,这归因于离子通过晶体的平行通道的存在。软供体配体位点(DMF中的羰基氧或吡啶中的芳族氮)与硬离子的错配导致无障碍,即温度独立性离子迁移。使用两种常见软-碱供体溶剂来实现相似结果表明这种方法对于形成软固体电解质材料的通用性。
所有操作均在干燥N2或氩气氛围下使用严格排除水的Schlenk和手套箱技术进行。使用从化学供应商(Aldrich或Strem)购买的无水LiCl。DMF和吡啶在N2氛围下使用CaH2作为清除剂进行蒸馏。Et2O使用二苯甲酮羰游基钠作为清除剂进行蒸馏。所有溶剂在使用前12h存储于手套箱中的活化分子筛上。单晶和粉末衍射在Bruker APEX II DUO衍射仪上进行。Mo Kα辐射用于单晶结构确定,而Cu Kα辐射用于粉末衍射研究。通过AC阻抗光谱使用Gamry Interface 1000稳压器/稳流器/ZRA在频率范围10-100kHz中测量离子电导率。使用不锈钢阻挡电极获得从-78至20℃的温度依赖性电导率,所述电极在气相色谱烘箱中受到温度控制。在各温度下,所述电池平衡30min并且对冷却和加热循环的电导率(σ)测量值相同。所报道的电导率来自斜线阻抗数据与等效电路的拟合以推断出体积电阻(R)。比离子电导率(σ)获自σ=t/AR,其中R为体积电阻(Ω),A=所述不锈钢电极的面积(1cm2),并且t=电解质材料的厚度。热重分析(TGA)在TA仪器Hi-Res TGA 2950上以10℃/min的攀升速率并且在N2吹扫气体下获得。傅里叶变换红外光谱(FTIR)数据在具有Specac Golden Gate ATR附件(菱形透镜)的Nicolet 580研究FTIR上获得,所述附件配备有具有100次扫描和4cm-1分辨率的MCT检测器。
合成
通过使无水LiCl溶解于DMF或吡啶中获得单晶。所述晶体通过在双瓶装置中在室温下使Et2O蒸气扩散至这一溶液中来生长。
使用相同方案制备DMF:LiCl和py:LiCl的整装粉末样品。0.2-0.5g(5.2mmol)LiCl在搅拌下溶解于DMF或吡啶(每0.1g LiCl为2mL溶剂)中直至溶解。溶解为缓慢的,典型地耗时约20分钟。所述溶液用二乙醚缓慢地稀释(以体积计1∶4)并且所述溶液混合约5分钟,产生白色沉淀物。过滤所述混合物,并且固体用二乙醚洗涤数次,并且在真空下干燥。所述化合物存储于手套箱冷冻器中的胶带密封闪烁瓶中。基于LiCl的产率:DMF:LiCl,87%。py:LiCl,84%。熔点DMF:LiCl70℃(分解),py:LiCl 65℃(分解)。
结果
单晶X射线衍射识别所述固体为LiCl与相应溶剂的分子的1∶1加合物。参看图6,所述结构示出通过锂原子与DMF和吡啶配体的软路易斯供体(分别为O和N)之间的微弱路易斯酸/碱加合物与所述晶格相互作用的1-D离子键结Li2Cl2菱面体的形成。参看图7,在DMF:LiCl的固态AT-IR光谱中的红移C=O伸展(1640cm-1)确认了锂离子与软羰基氧供体之间C=O--Li接触的存在。菱面体基序进一步通过相邻等效配体通过CH-Cl氢键(未示,)相互作用。CH-Cl氢键不常见,但先前已有报道(Aakeroy等人,1999,New J of Chemistry 23,145-152)。
尽管有类似的Li2Cl2菱面体结构,这些单元的3D堆积在两种加合物中不同(参看图6)。DMF:LiCl晶体展现通过锂原子端对端安排的交替的Li-Cl和Li-O(DMF)菱面体的线性安排,使得近间隔锂原子的链平行于晶胞的a轴存在。氯离子通道因此也平行于a轴定向,但这些离子之间的间隔为两倍大。在py:LiCl结构中,Li2Cl2单元再次沿着晶胞的a轴以Z字形模式堆积成边缘熔合的菱面体。这将锂离子跨所述菱面体的面以的距离,并且距相邻菱面体中的最近Li以的距离放置。
与迄今所报道的软固体电解质(Fergus,2010,J of Power Sources 195,4554-4569)相比,本发明的组合物展现优秀电导率。另外,这些组合物针对所测量的AC电导率极具温度独立性,所述AC电导率在20℃下展示~10-4S/cm的值(图8)。这种低温依赖性与具有低活化障碍(在室温区域中为14.2kJ/mol)的离子迁移事件一致。基于在电导率测量之前和之后的粉末衍射研究,化合物身份和结晶度在测量过程中充分维持(参看图9和10)。
对DMF-LiCl和Py-LiCl的热重分析(TGA)指示所述盐-基质共晶体的热稳定性随着基质分子的增加的沸点(Tb)而增加。TGA曲线(参看图11和12)示出降解温度略微低于纯液体的Tb(DMF的Tb=153℃,Py=115℃)。这可归因于由于在基质分子从固体晶格(U晶格(LiCl)=-829kJ/mol)升华时由固体溶液相Li+和Cl-离子形成纯LiCl(s)而产生的放热,其相对于纯液体的Tb降低了降解温度。
关于Py-LiCl的晶体结构报告
大致尺寸0.045mm×0.090mm×0.250mm的C5H5ClLiN样本用于X射线结晶分析。测量X射线强度数据。
使用斜方晶晶胞的数据整合产生了对最大θ角28.06°(分辨率)的总计2313次反射,其中1328次为独立的(平均冗余度1.742,完全性=98.5%,Rint=2.22%)并且1220次(91.87%)大于2σ(F2)。最终晶胞常数体积=是基于超出20σ(I)的反射的XYZ-质心的细化。所计算的最小和最大透射系数(基于晶体大小)为0.5464和0.7456。
所述结构使用Bruker SHELXTL软件包,使用空间群P 21 21 21进行解析并且细化,其中关于式单位C5H5ClLiN,Z=4。使用74个变量对F2进行的最终各向异性全矩阵最小平方细化在R1=2.93%(关于观察到的数据)和wR2=6.72%(关于所有数据)处会聚。拟合优度为1.058。最终差异电子密度合成中的最大峰为并且最大空穴为其中RMS偏差为基于最终模型,所计算的密度为1.335g/cm3并且F(000)为248e-。
Py-LiCl的热椭球曲线在图13中示出,并且相关数据概述于表1-5中。
表1.关于pyLiCl的样品和晶体数据。
表2.关于pyLiCl的数据收集和结构细化。
表3.关于pyLiCl的原子座标和等效各向同性原子位移参数
U(eq)是定义为正交化Uij张力器的迹线的三分之一。
表4.关于pyLiCl的键长
表5.关于pyLiCl的键角(°)。
关于DMFLiCl的晶体结构报告
大致尺寸0.055mm×0.132mm×0.255mm的C3H7ClLiNO样本用于X射线结晶分析。测量X射线强度数据。
使用单斜晶晶胞的数据整合产生了对最大θ角27.77°(分辨率)的总计4102次反射,其中1394次为独立的(平均冗余度2.943,完全性=99.8%,Rint=6.87%)并且862次(61.84%)大于2σ(F2)。最终晶胞常数β=98.826(6)°,是基于超出20σ(I)的反射的XYZ-质心的细化。所计算的最小和最大透射系数(基于晶体大小)为0.6103和0.7456。
所述结构使用Bruker SHELXTL软件包,使用空间群P 1 21/c 1进行解析并且细化,其中关于式单位C3H7ClLiNO,Z=4。使用66个变量对F2进行的最终各向异性全矩阵最小平方细化在R1=5.39%(关于观察到的数据)和wR2=12.43%(关于所有数据)处会聚。拟合优度为1.009。最终差异电子密度合成中的最大峰为并且最大空穴为其中RMS偏差为基于最终模型,所计算的密度为1.304g/cm3并且F(000)为240e-。
DMF-LiCl的热椭球曲线在图14中示出,并且相关数据概述于表6-10中。
表6.关于DMFLiCl的样品和晶体数据。
表7.关于DMFLiCl的数据收集和结构细化。
表8.关于DMFLiCl的原子座标和等效各向同性原子位移参数
U(eq)是定义为正交化Uij张力器的迹线的三分之一。
表9.关于DMFLiCl的键长
表10.关于DMFLiCl的键角(°)。
实施例2:二苯基甲烷-LiCl共晶体
制备二苯基甲烷(DPM)-LiCl共晶体组合物。二苯基甲烷(DPM)-LiCl共晶体的结构在图2中示出。这种结构代表盐于烃基质中的固体溶液的显著实例,所述基质具有用于Li+离子的交叉2-D通道(在44°下),但仅具有用于Cl-离子的非交叉通道。具有较高维度通道的晶体提供更大数目的用于离子迁移的路径,因为一维系统中的离子电导率可仅受通道中的一些杂质或缺陷限制(Kudo和Fueki,1990,Solid State Ionics,Kodansha Ltd;VCH:Tokyo;Weinheim)。尽管二苯基甲烷不具极性(如DMF和吡啶),所述芳烃为可聚合的,允许所述基质与Li和Cl离子之间的偶极-偶极相互作用,从而允许形成离子分子基质。DPM-LiCl晶体不展现与所述基质的显著离子-偶极相互作用,因为其位于与非极性C-H键相邻处。虽然这些C-H键必定极化并且提供与离子的偶极-偶极接触(Aakeroy等人,1999,New J of Chemistry 23,(2),145-152;Youm等人,2006,Polyhedron 25,(14),2717-2720),但所述基质对所述离子的亲和力有可能比对于DMF、Py或甘醇二甲醚系统中的杂原子的亲和力低得多(Zhanget等人,2007,Angewandte Chemie-International Edition,46,(16),2848-2850;Zhang等人,2007,J of the American Chemical Society 129,(28),8700-8701;Moriya等人,2012,Chemistry-a European J 18,(48),15305-15309),潜在地导致低障碍离子迁移。
实施例3:包含DMF-LiCl或Py-LiCl的薄膜的制备
通过将POSS-PEG8(POSS-PEG8的结构在图15中示出)溶解于二乙醚中并且使DMF-LiCl或Py-LiCl的晶体悬浮来制备固体膜,使得在二乙醚蒸发之后,存在10重量%POSS-PEG8。所有制剂均在Ar吹扫的手套箱中。在蒸发之后,所述膜在-78℃下为硬固体并且在-30℃下为橡胶状固体。DSC数据展示POSS-PEG8未结晶,并且Tg增加(指示Li+溶解度),并且关于纯样品展示相同的DMF-LiCl晶体结构。来自包含DMF-LiCl或Py-LiCl和POSS-PEG8的薄膜的分析的数据在图16(DSC曲线)和17(温度依赖性电导率曲线)中示出。
实施例4:共结晶LiCl·N,N-二甲基甲酰胺中的体相、低障碍离子导电
LiCl与N,N-二甲基甲酰胺(DMF)的共结晶产生具有组成LiCl·DMF的晶体。这些晶体在结晶a方向上形成线性、平行离子通道。通过电化学阻抗光谱进行的电导率测量产生1.3×10-4S/cm的值,所述值为关于有机固体电解质观察到的最高室温电导率。所述材料还在-15-25℃的环境温度区域中展现14kJ/mol的低活化障碍,和锂离子迁移数0.25。对在不同压力下制备的球粒和对含有导电粘合剂五环[9.5.1.13,9.15,15.17,13]八硅氧烷-聚乙二醇(POSS-PEG8)的样品进行的电导率测量指示在整体结晶相中存在导电,其中在晶粒边界处具有渗透LiCl/DMF液体层,表示当这一层冷冻时所测量的电阻率的主要来源。
材料的合成和一般实验程序与上文实验实施例1中关于LiCl·DMF所述的那些相同或相似。
微晶LiCl·DMF的经过压制的球粒展现与先前报道的其它软固体电解质相比优秀的电导率,其中在25℃下室温电导率为1.3×10-4S/cm(图8)。所测量的电导率具有小温度依赖性。在电导率曲线中看来存在两个线性区域,在25℃与-15℃之间在所述区域中产生14.2kJ/mol的阿伦尼乌斯活化能(参看下文中关于由电导率数据计算的活化能的进一步描述)并且在-25℃与-78℃之间产生48kJ/mol的阿伦尼乌斯活化能或在整个温度区间中产生45kJ/mol的阿伦尼乌斯活化能(图18),低于典型聚合物电解质值(在低温下50-100kJ/mol)并且类似于关于无机锂导体所测量的值(25-60kJ/mol(Knauth,2009,Solid State Ionics 180:911;Bron等人,2013,J.of the Am.Chem.Soc.2013,135:15694))。基于在电导率测量之前和之后的粉末衍射研究,化合物身份和结晶度在测量过程中充分维持(图19)。所述化合物在高温下降解(>40℃)。关于DMF·LiCl的热数据,即热重分析(TGA)(图12)指示,分解温度略微低于纯液体的Tb(DMF的Tvap=153℃,LiCl·DMF的Tmax为~140℃,但在较低温度下开始至实现)。虽然不希望受理论束缚,但这可归因于由于在基质分子从固体晶格(U晶格(LiCl)=-829kJ/mol)升华时由固体溶液相Li+和Cl-离子形成纯LiCl(s)而产生的放热,其相对于纯液体的Tb降低了降解温度。
关于LiCl·DMF的阻抗数据的尼奎斯特曲线由单一半圆和一个尖锋构成(图9),这可分别归因于组合的主体和晶粒边界电阻,和归因于离子阻挡电极的电极极化。在无机Li+导体的情况下,有时观察到两个半圆(Bron等人,2013,J.of the Am.Chem.Soc.2013,135:15694;Ohta等人,2011,J.of Power Sources 196:3342)(但并非始终,参看Kamaya等人,2011,Nature Materials 10:682),并且归因于主体和晶粒边界电阻。不同于其中使用高温退火(在~500℃下)来改进晶粒之间的接触的无机晶体的情况,所述方法不可能使用此处描述的共结晶样品。
为了评估晶粒边界电阻率的作用,在不同压力下并且使用不同干燥方案来制备样品球粒。在较高压力(1500psi)下制备的球粒产生具有改进的信噪比的数据。尼奎斯特曲线在低频率区域中具有较少噪声,因为用于制备所述球粒的压力增加,与由不良晶粒接触产生的显著电阻率一致。进一步地,过度干燥导致降低的电导率,表明液体样LiCl/DMF的边界层提供晶粒之间的接触。TGA数据(图12)示出三个重量损失步骤,虽然不希望受理论束缚,但所述步骤可归因于“主体”DMF(其在获得电导率数据之前从所述球粒去除(~50℃)),以及在“晶粒边界”层(~120℃)中和在晶粒本身(~140℃)中的DMF。关于塑性结晶盐-锂盐混合物的电导率增加,并且另外关于塑性结晶溶剂-锂盐混合物和结晶聚合物电解质,已经提出在晶粒边界处渗透液体混合盐相结构域而非固态扩散的相似机制(Henderson等人,2012,Advanced Energy Materials 2:1343)。
晶粒边界的作用的第二种测试是制备具有10%的导电粘合剂五环[9.5.1.13,9.15,15.17,13]八硅氧烷-聚乙二醇(POSS-PEG8)的球粒。这种材料的电导率曲线(图18)在POSS-PEG8的玻璃转变温度(Tg)以上展现基本上与无粘合剂样品相同的电导率值(图12),指示这种液体样层促进晶粒之间的导电。然而,POSS-PEG8结合的样品的电导率在POSS-PEG8的玻璃转变温度下急剧降低,表明在晶粒边界处的玻璃状材料限制电导率。类似地,1M LiCl/DMF溶液(图18)与LiCl·DMF晶体的电导率曲线的比较指示在~-70℃下电导率的降低是归因于在晶粒边界处液相的固化(图12)。虽然不希望受理论束缚,但这些结果表明虽然离子的导电是通过主体结晶相中的LiCl·DMF,但在晶粒边界处可出现显著电阻率。
基于使用Li°(s)电极的阻抗测量,锂离子迁移数(tLi+)为0.25(图9)。使用Li°的界面电阻由于锂金属与DMF的已知反应性(Butler和Synnott,1970,J.of the Am.Chem.Soc.,92:2602)而较高(图7)。尽管DMF已经单独(Croce等人,1996,J.of the Electrochem.Soc.,143:154)、在混合溶剂中(Tobishima等人,1988,Electrochimica Aeta,33:239)并且在聚合物凝胶电解质(Voice等人,1994,Polymer 35:3363)中关于锂离子电池的电导率研究进行了研究,其由于在电极表面反应性和钝化方面的问题而不用于实际应用(Advances in Lithium-Ion Batteries;Springer US,2002)。
由电导率数据计算的活化能
活化能(Ea)使用阿伦尼乌斯方程在两个不同温度区域中计算。在高温区域(25℃至-15℃)中,Ea=14.2kJ/mol,在低温区域(-20℃至-60℃)中,Ea=48kJ/mol,并且在整个温度范围(25℃至-60℃)内,Ea=45kJ/mol。均小于在温度区间0至50℃内关于由[CH3O(CH2CH2O)nCH3]:LiAsF6(n=3,4)制得的结晶固体甘醇二甲醚复合物获得的所述数据,其中Ea=55kJ/mol(n=3)或68kJ/mol(n=4)。不希望受理论束缚,log(σ)对1/T的线性变化表明导电机制为固定位点之间的离子跳跃,而非最好通过VTF方程描述的在离子与聚合物骨架移动之间的耦合动力学(Zhang等人,2007,J.of the Am.Chem.Soc.,129(28):第8700-8701页).
迁移数
在15℃下使用对称锂阻挡电极测量DMF:LiCl的迁移数(图20)。由于DMF对锂金属具腐蚀性,10%POSS-PEG作为粘合剂添加以限制DMF与锂的接触。使用由Vincent和合作者开发的方法(Evans等人,1987,Polymer,28(13):第2324-8页)如下计算迁移数(Appetecchi等人,1995,Electrochimica Acta,40(8):第991-997页;Heo等人,2004,Electrochimica Acta,50(2-3):第345-349页):
tLi+=Iss(ΔV-IoRo)/Io(ΔV-IssRss)
在15℃下使电池平衡几乎三小时之后,通过电化学阻抗光谱(EIS)测量归因于阴离子和阳离子的初始电阻Ro和电流Io,并且接着施加DC脉冲(ΔV=30mv)以极化所述电池。获得随时间而变的电流,并且测量现在仅归因于阳离子的稳态电阻(Rss)和电流(Iss)。此处Ro和Rss为钝化层的界面电阻,所述钝化层即在锂电极与具有10%POSS-PEG8的DMF:LiCl之间形成的固体电解质界面。
为了在锂离子电池组中使用电解质,可需要高锂离子迁移数,因为其降低电池极化。关于具有10%POSS-PEG8的DMF:LiCl,tLi+=0.28±0.03,高于多种商业锂离子电池电解质(Xu,2004,Chemical Reviews,104(10):4303-4418)和聚合物电解质(Agrawal和Pandey,2008,J.of Physics D:Applied Physics,41(22):223001),但仍指示大多数电流由Cl-离子携带(图21A和21B)。
界面电阻
在各循环中,锂损失并且在电解质与阳极(锂)之间形成界面,所述界面被称作固体电解质界面(SEI)层。关于具有10%POSS-PEG8的DMF:LiCl,界面电阻增加持续几乎6小时并且接着降低持续50小时(图20C)并且接着保持恒定(测试持续另外100小时)。在使氩气吹扫的手套箱中的电池开路时,锂金属看起来颜色深(并非最初的发光、金属光泽),指示电解质与Li°金属反应,并且是界面电阻从40kΩ至80kΩ大幅增加的原因。
本文所引用的每篇专利、专利申请和公布的公开内容均以引用的方式整体并入本文中。虽然本发明已参考具体实施方案加以公开,但显而易见的是,本发明的其它实施方案和变化形式可由所属领域的技术人员不悖离本发明的真实精神和范畴来设计。随附权利要求书意图解释为包括所有所述实施方案和相等变化形式。
- 还没有人留言评论。精彩留言会获得点赞!