靶向碳酸酐酶Ⅸ的磺胺类化合物、中间体及制备与应用的制作方法
本发明涉及磺胺类化合物,尤其是涉及一种靶向碳酸酐酶Ⅸ的磺胺类化合物、中间体及其制备方法。
背景技术:
碳酸酐酶Ⅸ(carbonic anhydrase Ⅸ,简称CA Ⅸ)是碳酸酐酶家族的(CAs)的异构体之一。它是由酸性氨基酸组成的跨膜糖蛋白,分布在细胞膜和细胞核,相对分子质量分别为58kD和54kD(Opavsky R.,Pastorekova S,Zelnik V,et al.Human MN/CA9gene,a novel member of the carbonic anhydrase family:Structure and exon to protein domain relationships[J].Genomics,1996,(3),480-487.Supuran C T,Scozzafava A.Carbonic anhydrase inhibitors and their therapeutic potential[J].Expert Opinion on Therapeutic Patents,2000,10(5),575-600.)。CA Ⅸ的主要功能是参与调节跨膜的离子和气体转运,同时还可能参与调节细胞增殖和转化,并与肿瘤的发生发展密切相关(Zhu Y,Zhou X Y,Yao X D,et al.Prognostic value of carbonic anhydrase IX expression in penile squamous cell carcinoma:A pilot study[J].Urologic Oncology-Seminars and Original Investigations,2013,31(5),706-711.Dungwa J V,Hunt L P,Ramani P.Carbonic anhydrase IX up-regulation is associated with adverse clinicopathologic and biologic factors in neuroblastomas[J].Human Pathology,2012,43(10),1651-1660.)。CA Ⅸ蛋白除在正常人消化道及其相关器官有表达外,在大多数正常人组织中几乎不表达。而CA Ⅸ在相应的肿瘤组织中过量表达,研究发现,CA Ⅸ在肾细胞癌、结直肠癌、宫颈癌、膀胱癌、非小细胞肺癌、肝癌、胰腺癌、卵巢癌、乳腺癌等表达增加(Takacova M,Bartosova M,Skvarkova L,et al.Carbonic anhydrase IX is a clinically significant tissue and serum biomarker associated with renal cell carcinoma[J].Oncology Letters,2013,5(1),191-197.Pirincci N,Gecit I,Gunes M,et al.Serum adenosine deaminase,catalase and carbonic anhydrase activities in patients with bladder cancer[J].Clinics,2012,67(12),1443-1446.Yu S J,Yoon J H,Lee J H,et al.Inhibition of hypoxia-inducible carbonic anhydrase-IX enhances hexokinase II inhibitor-induced hepatocellular carcinoma cell apoptosis[J].Acta Pharmacologica Sinica,2011,32(7),912-920.Schutze D,Milde-Langosch K,Witzel I,et al.Relevance of cellular and serum carbonic anhydrase IX in primary breast cancer [J].Journal of Cancer Research and Clinical Oncology,2013,139(5),747-754.)。CA Ⅸ是肿瘤特异性的相关蛋白,因而它是极具潜力的作为肿瘤靶向治疗和分子影像诊断的靶标。
正电子发射断层显像(Positron Emission Tomography,PET)技术具有定量分析、高灵敏度、高空间分辨率、全身三维影像等特点,在肿瘤、脑神经系统疾病、心脏疾病等的研究及临床应用中发挥着越来越重要的作用。而在PET显像的常用的核素中,F-18半衰期相对较长,对组织有较低的辐射剂量和较短的射程,是最常用的正电子核素,适于复杂的PET药物的合成和临床应用。
目前,磺胺类化合物是临床上使用的最为重要的一种CA Ⅸ抑制剂。通过对磺胺类的CA Ⅸ抑制剂进行F-18,发展一种特异性的PET探针,用于对CA Ⅸ过量表达的肿瘤的早期诊断和预后评估,具有十分重要的应用前景。但是,关于靶向CA Ⅸ的磺胺类的18F标记PET探针的相关研究国内外均鲜有报道,使得这一发明具有很好的创新性和较好应用价值。
技术实现要素:
本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种靶向碳酸酐酶Ⅸ的磺胺类化合物、中间体及制备与应用。
本发明的目的可以通过以下技术方案来实现:
本发明第一方面提供一种靶向碳酸酐酶Ⅸ的磺胺类化合物,以及靶向碳酸酐酶Ⅸ的磺胺类化合物的参比化合物。靶向碳酸酐酶Ⅸ的磺胺类化合物以及参比化合物的结构式分别下式A和A’所示
其中,n=0~6,较佳的为0,1和2,R1为H、C1~C4的直链烷基、C3~C4支链烷基、C3~C6环烷基或苯基-C1~C4烷基。
其中,所述的C1~C4的直链烷基为如下任一基团:
其中n=1~4,较佳的为1和2;
所述的C3~C4支链烷基为如下任一基团:
所述的C3~C6环烷基为如下任一基团:
其中n=1~4;
所述的苯基-C1~C4烷基为如下任一基团:
其中n=1~4。
本发明中,当F为18F时,所述的磺胺类化合物为18F标记磺胺类化合物,即化合物A。当F为普通的氟元素时,所述的磺胺类化合物为参比化合物A’。
本发明第二方面提供一种靶向碳酸酐酶Ⅸ的磺胺类化合物中间体,其结构式如下:
其中,n=0~6,R1为H、C1~C4的直链烷基、C3~C4支链烷基、C3~C6环烷基或苯基-C1~C4烷基,R1的具体选择同化合物A,R2为亲核取代反应中常见的离去基团,选自以下基团:
NO2、I-、Br-或Cl-。
本发明第三方面提供靶向碳酸酐酶Ⅸ的磺胺类化合物(化合物A)、其参比化合物A’以及靶向碳酸酐酶Ⅸ的磺胺类化合物中间体的制备方法。
将化合物B与18F-进行亲核取代反应,即制得靶向碳酸酐酶Ⅸ的磺胺类化合物 (化合物A)。
其中,n与R1与靶向碳酸酐酶Ⅸ的磺胺类化合物中的定义相同,R2与靶向碳酸酐酶Ⅸ的磺胺类化合物中间体中定义相同。
本发明中,所述的亲核取代反应的方法和条件可为有机合成领域此类反应中所用的方法和条件,进行亲核取代反应时,相转移催化剂为环状冠醚类催化剂或季铵盐类催化剂,所述的环状冠醚类催化剂选自18冠6或4,7,13,16,21,24-六氧-1,10-二氮双环[8.8.8]二十六烷(简写为K222),所述的季铵盐类催化剂选自碳酸氢四丁基铵或氢氧化四丁基铵。当使用环状冠醚类相转移催化剂时,反应中还需加入常用的钾盐,主要为碳酸钾、碳酸氢钾、草酸钾等。
本发明经过大量实验,特别优选出下述方法和条件:有机溶剂中,惰性气体保护下,K222、钾盐和18F-的混合物与化合物B进行亲核取代反应,即可。
其中,所述的钾盐较佳地为草酸钾和碳酸氢钾,优选草酸钾。所述的溶剂较佳地为无水乙腈、无水四氢呋喃、无水DMF(N,N-二甲基甲酰胺)和无水DMSO(二甲基亚砜)中的一种或多种,优选无水DMF。化合物B在反应液中的浓度较佳的为10~500mmol/L,更佳的为50~100mmol/L。所述的K222和草酸钾的摩尔比较佳的为1:5~10:1,更佳的为1:2~3:1。18F-的活度较佳的为50μCi~5Ci,更佳的为10mCi~1Ci。K222和化合物B的质量比较佳的为1:4~10:1,更佳的为2:1-5:1。所述的惰性气体较佳的为氮气和/或氩气和/或氦气。所述的亲核取代反应的温度较佳的为60~200℃,更佳的为90~160℃。所述的亲核取代反应可以在很短的时间内完成,如1~80分钟,更佳的为5~30分钟。
所述的含有K222、草酸钾和18F-的混合物可通过下述方法制得:用K222(即Kryptofix 222)溶液淋洗富集18F-的QMA柱,蒸干溶剂,即可。
其中,K222溶液可通过下述方法制得:将K222,草酸钾,乙腈和水配成溶液,即可。其中,各成分含量范围如下:每1mL乙腈中,有30~120μL水,1~10mg草酸钾,5~25mg K222。配置的方法可以为向1mL乙腈中加入30~140μL水,1~10mg草酸钾,5~25mg K222,即可。最常用的一种配比为:每960μL乙腈中,有 15mg K222,4.0mg草酸钾,40μL水,配成溶液即可。
上述亲核取代反应完成后,可用本领域常规的后处理和提纯方法进行提纯。本发明优选用放射性HPLC分离纯化标记产物,用放射性HPLC分离纯化之前,也可先用Sep-Pak C18柱对产物进行纯化。
将HPLC分离纯化得到的溶液减压浓缩,再溶于乙醇和生理盐水的混合溶液(乙醇含量<10%),经无菌滤膜过滤,得到18F标记制剂。
本发明中,当R2为NO2时,所述的标记前体化合物B可由下列方法制得,当R2为其他离去基团时,制备方法相似。
其中,n和R1的定义同前所述。
其中,所述的反应的方法和条件可为本领域此类反应的常规方法和条件,本发明特别优选下述方法和条件:
在乙腈、1-乙基-3-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC.HCl)、三乙胺(TEA)的混合物中,将化合物C与D进行反应,即可。其中,所述的反应的温度较佳地为5~50℃。所述的化合物C:D:EDC.HCl:TEA=1:(1~3):(1~5):(1~8)。该反应完成后,可用本领域常规的后处理和提纯方法进行提纯。本发明优选下述提纯方法和条件:反应液减压浓缩除掉大部分溶剂,加入硅胶浓缩干后,柱层析纯化(展开剂:石油醚,乙酸乙酯)。
其中,当n=0时,化合物Cn(即C0)可由下列方法制得:
其中,所述的反应的方法和条件可为本领域此类反应的常规方法和条件,本发明特别优选下述方法和条件:
于E的浓硫酸溶液中,缓慢滴加浓硝酸与浓硫酸的混酸溶液,滴加完毕反应即可。其中,所述的滴加浓硝酸与浓硫酸的混酸溶液的温度较佳的为-25~5℃,所 述的滴加完毕后反应温度较佳的为5~50℃。所述的化合物E:HNO3=1:(1~10),所述的混酸体积比HNO3:H2SO4=1:(0.5~10)。该反应完成后,可用本领域常规的后处理和提纯方法进行提纯。本发明优选下述提纯方法和条件:柱层析纯化,展开剂为石油醚和乙酸乙酯。
当n=1~6时,化合物Cn可由下列方法制得:
其中,所述的反应的方法和条件可为本领域此类反应的常规方法和条件,本发明特别优选下述方法和条件:
C0的制备同前所述。
F的制备方法如下所述:
将C0于二氯亚砜(SO2Cl2)中反应即可。其中,所述的反应的温度较佳地为25~100℃。该反应完成后,可用本领域常规的后处理和提纯方法进行提纯。本发明优选下述提纯方法和条件:反应溶液冷却后减压浓缩即可。
G的制备方法如下所述:
在乙醚、氧化钙(CaO)、化合物F的混合物中,缓慢滴加CH2N2的乙醚溶液进行反应,即可。其中,所述的反应温度较佳地为-20~5℃。所述的化合物F:CH2N2:CaO=1:(1~10):(1~10)。该反应完成后,可用本领域常规的后处理和提纯方法进行提纯。本发明优选下述提纯方法和条件:反应混合物减压浓缩掉溶剂后,硅胶柱层析纯化(展开剂:正己烷/乙酸乙酯)。
C1的制备方法如下所述:
在化合物G的二氧六环溶液中,加入氧化银的催化溶液进行反应,即可。其中,所述的反应的温度较佳地为25~100℃,所述的化合物G:氧化银=1:(1~5)。所述的氧化银的催化溶液可由下述方法制备:硝酸银和氢氧化钠溶于硫代硫酸钠的水溶液即可(硝酸银:氢氧化钠:硫代硫酸钠:水=1:1:2:250)。该反应完成后, 可用本领域常规的后处理和提纯方法进行提纯。本发明优选下述提纯方法和条件:浓缩除掉溶剂后,硅胶柱层析纯化(展开剂:石油醚/乙酸乙酯)。
化合物C(n=2~6)的制备只需重复C0→F→G→C1的制备过程即可。
本发明中,所述的参比化合物A’可由下列方法制得:
在乙腈、1-乙基-3-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC.HCl)、三乙胺(TEA)的混合物中,将化合物C’与D进行反应,即可。其中,所述的反应的温度较佳地为5~50℃。所述的化合物C:D:EDC.HCl:TEA=1:(1~3):(1~5):(1~8)。该反应完成后,可用本领域常规的后处理和提纯方法进行提纯。本发明优选下述提纯方法和条件:反应液减压浓缩除后硅胶柱层析纯化(展开剂:石油醚,乙酸乙酯)。
当n=1~6时,化合物C’可由下列方法制得:
F’的制备方法如下所述:
将C0’于二氯亚砜(SO2Cl2)中反应即可。其中,所述的反应的温度较佳地为25~100℃。该反应完成后,可用本领域常规的后处理和提纯方法进行提纯。本发明优选下述提纯方法和条件:反应溶液冷却后减压浓缩即可。
G’的制备方法如下所述:
在乙醚、氧化钙(CaO)、化合物F’的混合物中,缓慢滴加CH2N2的乙醚溶液进行反应,即可。其中,所述的反应温度较佳地为-20~5℃。所述的化合物F:CH2N2:CaO=1:(1~10):(1~10)。该反应完成后,可用本领域常规的后处理和提纯方法进行提纯。本发明优选下述提纯方法和条件:反应混合物减压浓缩掉溶剂后,硅胶柱层析纯化(展开剂:正己烷/乙酸乙酯)。
C1’的制备方法如下所述:
在化合物G’的二氧六环溶液中,加入氧化银的催化溶液进行反应,即可。其中,所述的反应的温度较佳地为25~100℃,所述的化合物G:氧化银=1:(1~5)。所述的氧化银的催化溶液可由下述方法制备:硝酸银和氢氧化钠溶于硫代硫酸钠的水溶液即可(硝酸银:氢氧化钠:硫代硫酸钠:水=1:1:2:250)。该反应完成后,可用本领域常规的后处理和提纯方法进行提纯。本发明优选下述提纯方法和条件:浓缩除掉溶剂后,硅胶柱层析纯化(展开剂:石油醚/乙酸乙酯)。
化合物C’(n=2~6)的制备只需重复C0’→F’→G’→C1’的制备过程即可。
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明的第四方面提供一种靶向碳酸酐酶Ⅸ的磺胺类化合物的应用,本发明的靶向碳酸酐酶Ⅸ的磺胺类化合物用作碳酸酐酶Ⅸ的抑制剂,作为一种特异性PET显像探针,用于对CA Ⅸ过量表达的肿瘤的早期诊断和预后评估。
与现有技术相比,本发明具有以下优点及有益效果:
1、本发明中用于放射性标记的前体合成步骤简单,产率高,易于大量制备。
2、本发明的靶向碳酸酐酶Ⅸ的磺胺类化合物制备方法简单,反应时间短,更加适合放射药物临床应用,制备得到的18F标记化合物放射化学纯度大于99%。
3、本发明中所用到的试剂均为商品化试剂,原料方便易得。
附图说明
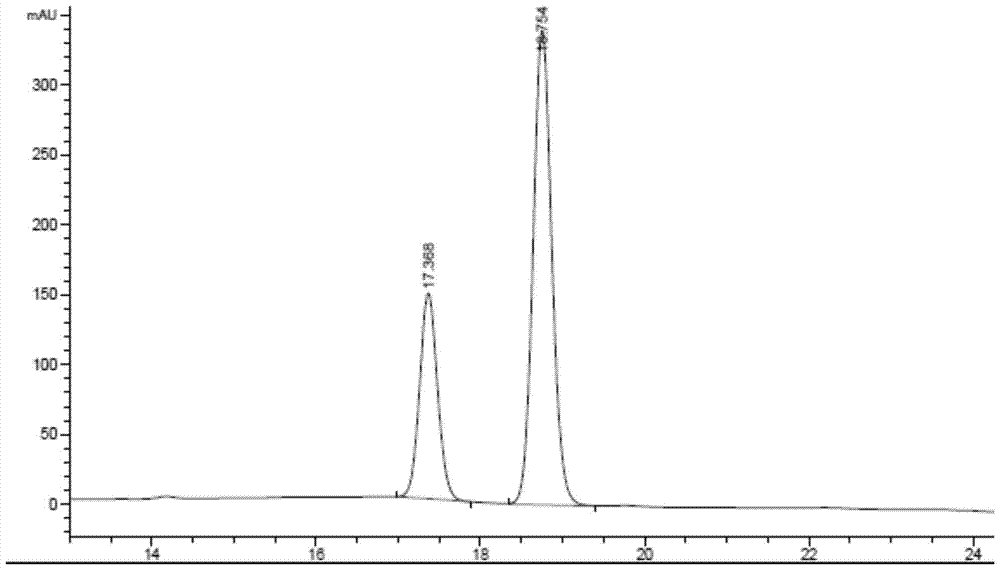
图1为实施例6的例一的标记产物A0与其参比化合物A0’(实施例5)共同进样的HPLC检测谱图。
图2为实施例5中的参比化合物A0’与实施例3的标记前体化合物B0一起进样的HPLC谱图。
具体实施方式
下面结合附图和具体实施例对本发明进行详细说明。
实施例1
5-硝基-2,3,6三氟苯甲酸(C0)的合成
冰浴下,2,3,6三氟苯甲酸(E)4.0g溶于30mL的浓硫酸中,0℃以下缓慢滴加浓硝酸(5.1mL)与浓硫酸(5.1mL)混酸溶液,滴加完毕后室温反应5h。 冰浴冷却上述反应液,搅拌下缓慢滴加冰水60mL。乙酸乙酯100mL/次萃取两次,合并乙酸乙酯层,饱和氯化钠溶液洗两次,有机层无水硫酸钠干燥后,减压浓缩,得淡黄色固体4.4g。收率为87.6%,纯度>98%(HPLC)。
C0的鉴定数据如下:TOF-ESI-MS:M(C7H2F3NO4)=221.09(m/z),244.0[M+Na]+,276[M+Na+CH3OH]+.
1H-NMR(300MHz,CDCl3)δ8.18(m,1H,H)
实施例2
5-硝基-2,3,6三氟苯乙酸(C1)的合成
1.室温下,235.7mg(1.07mmol)的C0溶于5mL的SOCl2中,80℃回流反应5h,反应液减压浓缩得到淡黄色液体F,直接用于下一步反应。
2.上述1中制得的F溶于5mL的乙醚中,加入CaO 120mg(2.14mmol),冰浴下缓慢滴加CH2N2(2.14mmol)的乙醚溶液进行反应。反应结束后,产品用硅胶柱层析纯化(展开剂:正己烷,乙酸乙酯)。得产品G 233.7mg,收率为89.1%,纯度99%(HPLC)。
3.化合物G 123.0mg(501.8umol)溶于5mL二氧六环溶液中,加入通过硝酸银170mg(1mmol)和氢氧化钠40mg(1mmol)溶于硫代硫酸钠316mg(2mmol)的水溶液9mL(500mmol)制得的氧化银的催化溶液,50℃加热进行反应。反应结束后,产品用硅胶柱层析纯化(展开剂:正己烷,乙酸乙酯)。得产品C1102.1mg,收率为86.5%,纯度99%(HPLC)。
C1的鉴定数据如下:TOF-ESI-MS:M(C8H4F3NO4)=235.01(m/z),258.0[M+Na]+.
1H-NMR(300MHz,CDCl3):8.75(s,1H,H),8.15(m,1H,CH),3.74(s,2H,CH2).
实施例3
标记前体化合物B0的制备
间氨基苯磺酰胺68.9mg(0.4mmol),C0 88.5mg(0.4mmol)溶于6mL乙腈中,加入EDC.HCl 76.7mg(0.4mmol)、三乙胺56uL(0.4mmol)于室温搅拌反应。反应结束后,产品用硅胶柱层析纯化(展开剂:正己烷,乙酸乙酯)。得产品B0125.2mg,收率为83.4%,纯度99%(HPLC)。
B0的鉴定数据如下:TOF-ESI-MS:M(C13H8F3N3O5S)=375.28(m/z),398.0[M+Na]+,430.0[M+Na+CH3OH]+.
1H-NMR(300MHz,CDCl3):δ11.36(s,1H,NH),8.67(m,1H,CH),8.23(s,1H,CH),7.75(m,1H,CH),7.59(m,2H,CH),7.43(s,2H,NH2).
实施例4
2,3,5,6-四氟苯乙酸(C1)的合成
1.室温下,194.1mg(1.0mmol)的C0’溶于5mL的SOCl2中,80℃回流反应5h,反应液减压浓缩得到淡黄色液体F’,直接用于下一步反应。
2.上述1中制得的F’溶于5mL的乙醚中,加入CaO 112mg(2.0mmol),冰浴下缓慢滴加CH2N2(2.0mmol)的乙醚溶液进行反应。反应结束后,产品用硅胶柱层析纯化(展开剂:正己烷,乙酸乙酯)。得产品G’193.9mg,收率为88.9%,纯度99%(HPLC)。
3.化合物G’109.1mg(0.5mmol)溶于5mL二氧六环溶液中,加入通过硝酸银170mg(1mmol)和氢氧化钠40mg(1mmol)溶于硫代硫酸钠316mg(2mmol)的水溶液9mL(500mmol)制得的氧化银的催化溶液,50℃加热进行反应。反应结束后,产品用硅胶柱层析纯化(展开剂:正己烷,乙酸乙酯)。得产品C1’96.0mg,收率为92.3%,纯度99%(HPLC)。
C1’的鉴定数据如下:TOF-ESI-MS:M(C8H4F4O2)=208.11(m/z),231.1[M+Na]+,263.1[M+Na+CH3OH]+.
1H-NMR(300MHz,CDCl3):δ10.1(s,1H,H),6.72(m,1H,CH),3.71(s,2H,CH2).
实施例5
参比化合物A0’的制备
间氨基苯磺酰胺34.5mg(0.2mmol),C0’38.8mg(0.2mmol)溶于6mL乙腈中,加入EDC.HCl 57.5mg(0.3mmol)、三乙胺42uL(0.3mmol)于室温搅拌反应。反应结束后,产品用硅胶柱层析纯化(展开剂:石油醚,乙酸乙酯)。得产品B0’57.6mg,收率为82.7%,纯度99%(HPLC)。
A0’的鉴定数据如下:TOF-ESI-MS:M(C13H8F4N2O3S)=348.27(m/z),371.0[M+Na]+,403.0[M+Na+CH3OH]+.
1H-NMR(300MHz,CDCl3):δ11.27(s,1H,NH),8.24(s,1H,CH),8.09(m,1H,CH),7.75(m,1H,CH),7.58(m,2H,CH),7.41(s,2H,NH2).
实施例6
18F标记PET探针N-(间磺胺基苯基)-2,3,6-三氟-5-[18F]氟-苯甲酰胺A0的制备
【例1】20mCi 18F-被季铵型阴离子柱QMA(美国Waters公司产品)捕获后,取1.0mL K222(即Kryptofix 222)溶液(15.0mg K222,5.1mg二水合草酸钾,1000μL乙腈,40μL水配成的溶液)将18F-冲洗到反应瓶中,反应瓶浸入100℃的油浴,氮气吹干,然后再加入500μL无水乙腈吹干,重复上述操作两次;将标记前体化合物B0(5mg)的500μL无水DMF溶液,氮气保护下迅速加入反应瓶,100℃下密闭反应10min,停止反应,冰水浴冷却,标记率为35%。
例一的标记产物A0与其参比化合物A0’(实施例5)共同进样的HPLC检测谱图如图1所示,HPLC条件:分析柱Agilent C18column(5μm,4.6mm×250mm)。流动相为含0.05%TFA水溶液(X)和含0.05%TFA的乙腈(Y),梯度分离条件为:0→10→15→30min,5%→5%→50%→50%Y。流速为1.0mL/min。经过UV(254nm)检测和放射性检测。
标记产物用HPLC检测并分离,分离后放化纯度>99%。减压浓缩除掉溶剂,用含8%乙醇的0.9%的医用生理盐水溶解,无菌滤膜过滤后得到可注射用PET探针。
【例2】50mCi 18F-被季铵型阴离子柱QMA(美国Waters公司产品)捕获后,取1.0mL K222(即Kryptofix 222)溶液(20.0mg K222,13.5mg二水合草酸钾,1000μL乙腈,50μL水配成的溶液)将18F-冲洗到反应瓶中,反应瓶浸入150℃的油浴,氮气吹干,然后再加入500μL无水乙腈吹干,重复上述操作两次;将标记前体化合物B0(10mg)的800μL无水DMF溶液,氮气保护下迅速加入反应瓶,150℃下密闭反应5min,停止反应,冰水浴冷却,标记率为38%。
实施例5中的参比化合物A0’与实施例3的标记前体化合物B0一起进样的HPLC谱图如图2所示,标记产物用HPLC检测并分离(美国Agilent 1100HPLC系统,半制备柱为Agilent C18column(9.3mm×250mm)。流动相为含0.05%TFA水溶液(X)和含0.05%TFA的乙腈(Y),梯度分离条件为:0-5-30-30.01-37-37.01-40min,30%→30%→40%→100%→100%→30%→30%Y。流速为4.0mL/min。经过UV(254nm)检测和放射性检测。)收集产物峰(tR=17.3min),分离后放化纯度>99%。减压浓缩除掉溶剂,用含8%乙醇的0.9%的医用生理盐水溶解,无菌滤膜过滤后得到可注射用PET探针。
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 靶向PD-1的多肽及其应用的制造方法与工艺
- 靶向同位素生产系统的制造方法与工艺
- 在制备68GA‑螯合物官能化的靶向剂中用作金属抑制剂的单糖、二糖或多糖的制造方法与工艺
- 用于引导和靶向按需物质递送的纳米结构的载体的制造方法与工艺
- 通过PROSTRATIN靶向K‑RAS介导的信号转导通路和恶性肿瘤的制造方法与工艺
- 肿瘤靶向核磁共振/荧光双模态成像造影剂及其制备和应用的制造方法与工艺
- pH氧化还原双敏感的PAMAM靶向纳米给药载体及其制备方法与制造工艺
- 具有卵巢肿瘤靶向D构型多肽及其基因递释系统的制造方法与工艺
- 靶向survivin纳米抗体及其制备方法和应用与制造工艺
- 用于PSMA靶向的成像和放射疗法的金属/放射性金属标记的PSMA抑制物的制造方法与工艺
- 阻断UBE2M介导Neddylation修饰在肾细胞癌治疗中的应用的制造方法与工艺
- 无载体共组装肿瘤靶向抗癌纳米药物及其制备方法与应用与流程
- 一种用于判断靶向单克隆抗体治疗肿瘤疗效的预测分子的制造方法与工艺
- 一种靶向CD47的免疫检查点抑制剂药物组合物及其制备方法与流程
- 具有淋巴靶向功能的生物自组装纳米晶注射剂及制备方法与流程
- 用于检测肝癌的基因标志物及其用途的制造方法与工艺
- 特异性shRNA筛选及其靶向Ang‑2基因抑制肺癌细胞的验证方法与流程
- 一种多功能自动化生物细胞提取分离控制系统及采用该系统实现的细胞提取分离方法与流程
- 一种融合蛋白及光敏剂复合物及其制备方法和应用与流程
- 具有可视化和显示光斑边界功能的光动力治疗系统的制造方法与工艺
- 肿瘤靶向核磁共振/荧光双模态成像造影剂及其制备和应用的制造方法与工艺
- pH氧化还原双敏感的PAMAM靶向纳米给药载体及其制备方法与制造工艺
- 具有卵巢肿瘤靶向D构型多肽及其基因递释系统的制造方法与工艺
- 靶向survivin纳米抗体及其制备方法和应用与制造工艺
- 用于PSMA靶向的成像和放射疗法的金属/放射性金属标记的PSMA抑制物的制造方法与工艺
- 一种线粒体靶向的粘度荧光探针及其制备方法和应用与制造工艺
- 一种兼具荧光可视、磁靶向和pH敏多功能的复合空心微球药物载体及其制备方法与制造工艺
- 一种用于修饰微泡的多肽以及靶向GBM的药物制剂的制造方法与工艺
- 白果内酯作为脑靶向增效剂在制备防治脑肿瘤药物的应用的制造方法与工艺
- 机器人辅助精确靶向穿刺软组织目标柔性针夹紧固定装置的制造方法
- 一种用于判断靶向单克隆抗体治疗肿瘤疗效的预测分子的制造方法与工艺
- 一种检测肺癌相关基因热点突变的试剂盒及其应用的制造方法与工艺
- 一种为肺癌提供离体个体化药物测试的方法及培养基与流程
- 一种谷胱甘肽/超声双重刺激响应纳米聚合物囊泡、其制备方法及应用与流程
- 一种多重刺激响应交联型聚合物纳米水凝胶、其制备方法及应用与流程
- 一种靶向CD47的免疫检查点抑制剂药物组合物及其制备方法与流程
- 具有淋巴靶向功能的生物自组装纳米晶注射剂及制备方法与流程
- 用于检测肝癌的基因标志物及其用途的制造方法与工艺
- 特异性shRNA筛选及其靶向Ang‑2基因抑制肺癌细胞的验证方法与流程
- 一种具有靶向功能仿病毒结构高分子囊泡及其制备与应用的制造方法与工艺