敲除中心碳代谢基因以提高格尔登素发酵水平的方法与流程
本发明涉及生物工程,尤其是涉及一种敲除中心碳代谢基因以提高格尔登素发酵水平的方法。
背景技术:
放线菌是一类高GC含量的革兰氏阳性菌,目前已知抗生素中有60%以上是由放线菌合成。链霉菌是一类高等放线菌,具有强大的抗生素合成能力和复杂的形态分化,故一直受到人们的关注。聚酮类化合物是一类由放线菌产生的重要的次级代谢产物,其在抗肿瘤、抗寄生虫、抗生素领域都有极其重要的应用。前期的实验已经证明,聚酮类化合物的生物合成主要依赖I型、II型和III型聚酮生物合成基因簇,其主要使用的前体物是乙酰辅酶A、丙二酰辅酶A、甲基丙二酰辅酶A、乙基丙二酰辅酶A等等。而这些前体物质主要是由初级代谢即EMP途径、TCA途径中的中间产物经过几步反应而形成。
格尔登素(geldanamycin)属于I型聚酮类抗生素,主要由吸水链霉菌的一些亚种如S.hygroscopicus NRRL 3602、S.hygroscopicus JCM4427产生。格尔登素可以与细胞中的热击蛋白HSP90结合,阻碍其执行分子伴侣功能,抑制肿瘤细胞的分裂,因此有着强的抗肿瘤活性。除此之外,它还有一定的抗革兰氏阳性菌、真菌、疟疾、原生动物和寄生虫的活性。格尔登素虽然有强烈的抗肿瘤活性,但由于它对一般细胞也有毒性且在水中溶解性差,所以不能进行临床应用。目前它的类似物17-[2-(dimethylamino)ethyl]amino-17-demethoxygeldanamycin(17-DMAG)和17-allylamino-17-demethoxygeldanamycin(17-AAG)都在临床检定阶段。格尔登素也广泛应用在科研中对于热击蛋白HSP90的研究中。
格尔登素的生物合成是经起始单元AHBA合成、PKS组装和后修饰步骤这三部分完成的。格尔登素的生物合成基因簇最早在Streptomyces hygroscopicus NRRL 3602中被发现并在2003年被详细地注释。后来又在Streptomyces hygroscopicus 17997中被克隆出来。
本微生物代谢国家重点实验室在2015年完成了格尔登素产生菌吸水链霉素XM201(Streptomyces hygroscopicus XM201)的全基因组测序。通过基因组注释和代谢网络分析,我们发现XM201拥有庞大的基因组。它的中心碳代谢途径中,很多基因都具有多个拷贝。目前,我们已经知道前体物的供应会影响到聚酮链的合成,进而影响化合物的最终产量,很多研究通过过量表达相关前体延伸单元合成酶的基因,大大提高了化合物的产量,如红霉素、盐霉素等。然而这些生成延伸单元的前体如1,3-二磷酸甘油酸、乙酰辅酶A、琥珀酰辅酶A等的合成,则是依赖于中心碳代谢的代谢流分配。因此对于格尔登素的合成来说,通过失活中心碳代谢的一些基因,导致前体物质的积累可能是提高抗生素的产量的一种有效途径。为此本发明尝试找到通过失活中心碳代谢的相关基因来提高格尔登素产量的方法,为格尔登素工业化规模的扩大和生产成本的降低提供有效的参考。
技术实现要素:
本发明的目的,就是为了提供一种敲除中心碳代谢基因以提高格尔登素发酵水平的方法。
为了达到上述目的,本发明采用了以下技术方案:一种敲除中心碳代谢基因以提高格尔登素发酵水平的方法,是通过失活吸水链霉菌XM201基因组中编码pyk的ORF3547,以及编码sdh的ORF1011、ORF6607和ORF4917,导致中心碳代谢流量重排及相关前体积累,使格尔登素的发酵产量得以提高。
所述编码pyk的ORF3547的序列如SEQ ID NO.1所示。
所述编码sdh的ORF1011的序列如SEQ ID NO.2所示。
所述编码sdh的ORF6607的序列如SEQ ID NO.3所示。
所述编码sdh的ORF4917的序列如SEQ ID NO.4所示。
构建步骤如下:
第一步:设计并构建用于ORF3547失活的同源重组质粒载体I;
第二步:设计并构建用于ORF1011失活的同源重组质粒载体II;
第三步:设计并构建用于ORF6607失活的同源重组质粒载体III;
第四步:设计并构建用于ORF4917失活的同源重组质粒载体IV;
第五步:将构建得到的四个同源重组质粒载体通过接合转移导入受体菌吸水链霉菌XM201中进行同源重组;
第六步:通过对突变株的筛选和阿泊拉霉素抗性验证,并通过PCR产物片段大小的差异筛选得到ORF3547、ORF1011、ORF6607、ORF4917失活的突变株。
所述质粒载体I的构建方法是,在质粒pJTU1278的KpnI/XbaI位点插入来自ORF3547的0~750bp区域左侧2025bpPCR片段和来自ORF3547的0~750bp区域右侧2045bp PCR片段,得到的载体通过HindIII位点连入阿泊拉霉素抗性基因;所述质粒载体II的构建方法是,在质粒pJTU1278的KpnI/XbaI位点插入来自ORF1011的上游17bp~基因内677bp区域左侧2068bp PCR片段和来自ORF1011的上游17bp~基因内677bp区域右侧2049bp PCR片段,得到的载体通过HindIII位点连入阿泊拉霉素抗性基因;所述质粒载体III的构建方法是,在质粒pJTU1278的XbaI/HindIII位点插入来自ORF6607的4~624bp区域左侧2078bp PCR片段和来自ORF6607的4~624bp区域右侧2069bp PCR片段,得到的载体通过EcoRI位点连入阿泊拉霉素抗性基因;所述质粒载体IV的构建方法是,在质粒pJTU1278的XbaI/HindIII位点插入来自ORF4917的上游111bp~基因内部720bp区域左侧2144bpPCR片段和来自ORF4917的上游111bp~基因内部720bp区域右侧2176bp PCR片段,得到的载体通过EcoRI位点连入阿泊拉霉素抗性基因。
所述发酵是指将链霉菌孢子或者菌丝体接种在种子培养基中于30℃、220rpm的转速下培养36小时后转接发酵培养基发酵6天。
所述种子培养基含有质量体积比为1%的葡萄糖、1%的蛋白胨和0.5%的酵母提取物;所述发酵培养基含有质量体积比为3%的淀粉、7%的葡萄糖、4%的黄豆饼粉、0.3%的硫酸铵、1%的碳酸钙、0.001%的氯化钴和0.1%的大豆油。
种子培养物按15%的接种量转接于发酵培养基。
本发明通过在染色体上置换的方法,在基因ORF3547、ORF1011、ORF6607、ORF4917位置上引入基因突变,导致丙酮酸激酶、琥珀酸脱氢酶基因的失活。本发明所得到的工程菌株其格尔登素的产量大幅提高。
本发明通过改变中心碳代谢的代谢流分配,提高了格尔登素前体的供应以此来提高格尔登素发酵水平,最终格尔登素发酵产量最高提高了一倍,最终产量达到1.5g/L,通过本发明可显著提高格尔登素的发酵产量,同时使发酵成本大幅降低。
本发明所涉及的质粒pJTU1278已经在SCI数据库文献《He Y,Wang Z,Bai L,Liang J,Zhou X,Deng Z:Two pHZ1358derivative vectors for efficient gene knockout in Streptomyces.Journal of Microbiology and Biotechnology 2010,20(4):678-682》中公开。
附图说明
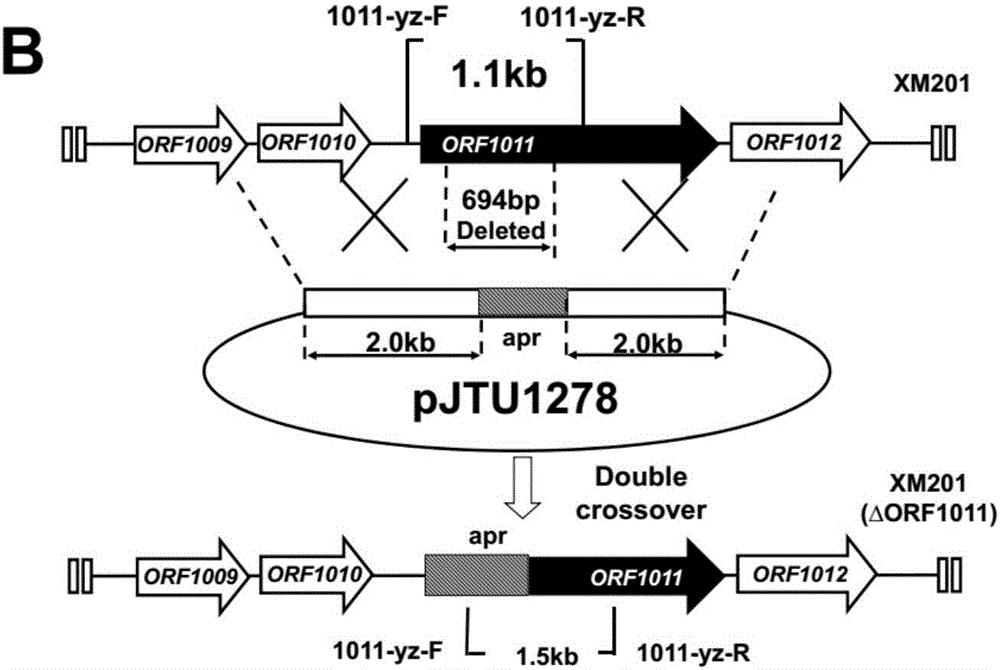
图1-4示意了基因ORF3547、ORF1011、ORF6607和ORF4917敲除的过程;
图5为基因1011、4917失活的突变株与野生型菌株XM201的前体物定量结果;
图6-7为基因ORF3547、ORF1011、ORF6607和ORF4917敲除后格尔登素发酵水平检测结果。
具体实施方式
下面通过实施例对本发明作进一步说明。本实施例在以本发明技术方案为前提下进行实施,并给出了详细的实施方式和过程,但本发明的保护范围不限于下述的实施例。下列实施例中未注明具体条件的实验方法,按照常规条件或制造厂商的建议条件。
实施例
步骤一:质粒pLQ610、pLQ611、pLQ612和pLQ613的构建
以吸水链霉菌XM201的基因组DNA为模板,分别使用两组引物3547L-F/R、3547R-F/R通过PCR扩增得到来自ORF3547的0~750bp区域左侧2025bp和右侧2045bp的同源臂,通过基因测序确认同源臂的正确性;在质粒pJTU1278的KpnI/XbaI位点插入来自ORF3547的0~750bp区域左侧2025bp PCR片段(KpnI/HindIII)和ORF3547的0~750bp区域右侧2045bpPCR片段(HindIII/XbaI);通过上述方法得到用于ORF3547基因失活的质粒pLQ610-1。在37℃水浴条件下,采用KpnI、HindIII和XbaI三种限制性内切酶进行酶切处理可以观察到2.0kb左右的两条重合的目标条带,表明质粒构建正确。将pLQ610-1用HindIII进行酶切后同阿泊拉霉素抗性基因进行连接,得到重组质粒pLQ610。
以吸水链霉菌XM201的基因组DNA为模板,分别使用两组引物1011L-F/R、1011R-F/R通过PCR扩增得到来自ORF1011的上游17bp~基因内677bp区域左侧2068bp和右侧2049bp的同源臂,通过基因测序确认同源臂的正确性;在质粒pJTU1278的KpnI/XbaI位点插入来自ORF1011的上游17bp~基因内677bp区域左侧2068bp PCR片段(HindIII/XbaI)和ORF1011的0~694bp区域右侧2049bp PCR片段(KpnI/HindIII);通过上述方法得到用于ORF1011基因失活的质粒pLQ611-1。在37℃水浴条件下,采用KpnI、HindIII和XbaI三种限制性内切酶进行酶切处理可以观察到2.0kb左右的两条重合的目标条带,表明质粒构建正确。将pLQ611-1用HindIII进行酶切后同阿泊拉霉素抗性基因进行连接,得到重组质粒pLQ611。
以吸水链霉菌XM201的基因组DNA为模板,分别使用两组引物6607L-F/R、6607R-F/R通过PCR扩增得到来自ORF6607的4~624bp区域左侧2078bp和右侧2069bp的同源臂,通过基因测序确认同源臂的正确性;在质粒pJTU1278的XbaI/HindIII位点插入来自ORF6607的3~624bp区域左侧2078bp PCR片段(XbaI/EcoRI)和ORF6607的4~624bp区域右侧2069bp PCR片段(EcoRI/HindIII);通过上述方法得到用于ORF6607基因失活的质粒pLQ612-1。在37℃水浴条件下,采用XbaI、HindIII和EcoRI三种限制性内切酶进行酶切处理可以观察到2.0kb左右的两条重合的目标条带,表明质粒构建正确。将pLQ612-1用EcoRI进行酶切后同阿泊拉霉素抗性基因进行连接,得到重组质粒pLQ612。
以吸水链霉菌XM201的基因组DNA为模板,分别使用两组引物4917L-F/R、4917R-F/R通过PCR扩增得到来自ORF4917的基因上游111bp~基因内部720bp区域左侧2144bp和右侧2176bp的同源臂,通过基因测序确认同源臂的正确性;在质粒pJTU1278的XbaI/HindIII位点插入来自ORF4917的基因上游111bp~基因内部720bp区域左侧2144bp PCR片段(XbaI/EcoRI)和ORF4917的基因上游111bp~基因内部720bp区域区域右侧2174bp PCR片段(EcoRI/HindIII);通过上述方法得到用于ORF4917基因失活的质粒pLQ613-1。在37℃水浴条件下,采用XbaI、HindIII和EcoRI三种限制性内切酶进行酶切处理可以观察到2.0kb左右的两条重合的目标条带,表明质粒构建正确。将pLQ613-1用EcoRI进行酶切后同阿泊拉霉素抗性基因进行连接,得到重组质粒pLQ613。
步骤二:将用于和染色体发生重组的质粒pLQ610、pLQ611、pLQ612和pLQ613导入野生型菌株XM201,并筛选得到ORF3547、ORF1011、ORF6607和ORF4917的部分缺失突变株,此突变株表征吸水链霉菌XM201的丙酮酸激酶或琥珀酸脱氢酶的部分失活。
图1-4示意了基因ORF3547、ORF1011、ORF6607和ORF4917敲除的过程。具体操作如下:将已构建完成用于基因缺失的质粒pLQ610、pLQ611、pLQ612和pLQ613转化进入宿主E.coli ET12567(含有pUZ8002质粒)中。取E.coli ET12567于含有Apr、Kan和Chl三种抗生素的LB中于37℃过夜培养,用相同的培养基,将过夜培养物按10%的比例转接一次并培养2.5小时,然后用新鲜的LB溶液漂洗菌体以除去培养物中的抗生素。于此同时制备野生型菌株XM201的新鲜孢子约109个,用TES溶液漂洗2~3次之后再用1mLTES溶液悬浮孢子,于50℃热激10min后再冷却至室温,等体积加入2×孢子预萌发培养液后于37℃培养0.5小时。将预萌发的孢子液与之前制备的宿主菌E.coli ET12567混合(孢子和宿主菌的比例约为10:1)均匀后涂布于SFM平板,待平板吹干后转移至30℃培养箱培养16小时后取出平板,分别取Apr和TMP两种抗生素的储存液40μL加入1mL无菌水中混匀后覆盖在SFM平板上,将平板晾干后转移至30℃培养箱中培养。一般5~7天后可见平板上有单菌落接合子长出,采用3547-yz-F/3547-yz-R、1011-yz-F/1011-yz-R、6607-yz-F/6607-yz-R和4917-yz-F/4917-yz-R为引物,通过菌丝体PCR和抗性验证的方法验证接合子正确后划线接种至SFM平板,30℃培养至产孢,收集孢子后按稀释107~108后接种至SFM平板培养2~3天可得到单菌。继续采用3547-yz-F/3547-yz-R、1011-yz-F/1011-yz-R、6607-yz-F/6607-yz-R和4917-yz-F/4917-yz-R为引物,通过菌丝体PCR的方法对单菌落筛选得到ORF3547、ORF1011、ORF6607和ORF4917缺失突变株。
上述步骤二中所用到的引物序列如表1所示:
表1
用于敲除的左右同源臂制备PCR反应体系和条件:DNA模板30ng,引物30pmol,50%DMSO 3μL,25mM Mg2+2μL,缓冲液3μL,KOD聚合酶1个单位,加纯水补齐至30μL;PCR条件:95℃5分钟;95℃30秒;60℃30秒;68℃2分钟;循环30次;68℃10分钟。
基因敲除的接合转移子和突变株筛选时的PCR体系和条件:DNA模板10~100ng,引物30pmol,50%DMSO 3μL,缓冲液3μL,Taq聚合酶0.5个单位,加纯水补齐至30μL;PCR条件:95℃5分钟;95℃30秒;60℃30秒;72℃1分钟;循环30次;72℃10分钟。
步骤三,丙酮酸激酶和琥珀酸脱氢酶基因失活菌株的发酵培养
种子培养基含有(质量体积比):葡萄糖1%,蛋白胨1%,酵母提取物0.5%,自然pH;
发酵培养基含有(质量体积比):淀粉3%,葡萄糖7%,黄豆饼粉4%,硫酸铵0.3%,碳酸钙1%,氯化钴0.001%,大豆油0.1%。调节pH=7.0~7.2,115℃灭菌30分钟。
步骤四,琥珀酸脱氢酶基因缺失突变株中前体物浓度的测定
收集24h的发酵产物,取20mL发酵液高速离心用无菌水洗涤两次,再用5mL无菌水重新悬浮,取1mL离心去上清后,加入1mL二苯胺试剂(1.5g二苯胺粉末溶于100mL冰醋酸中,加入1.5mL浓H2SO4,1mL1.6%的乙醛水溶液),60℃处理1小时,测定OD595,用核酸浓度表征菌体生物量。另取1mL菌体离心后加入1mL萃取液(甲醇:乙腈:0.1%乙酸水=45:45:10)重新悬浮,高速震荡破碎15分钟,间歇性的置于冰上冷却。15分钟之后高速离心取上清,经过0.22μm的有机相滤膜过滤之后可以用LC-MS/MS检测。
前体物辅酶A的检测使用的是LC-MS/MS的正离子模式下的多反应监测模式(Multiple Reaction Monitoring mode,MRM mode),分别选择的母分子和子分子分子量如下:丙二酰辅酶A(854>347),甲基丙二酰辅酶A(868>361),乙基丙二酰辅酶A(882>375)。色谱柱的参数:Agilent TC-C18,4.6×250mm,5μm;流动相流速为0.5mL/min;流动相:20mmol/L的乙酸铵水溶液(溶液A)和20mmol/L的乙酸铵甲醇溶液(溶液B);流动相变化:25%溶液B 5分钟,随后在10分钟内均匀达到100%溶液B,保持100%溶液B 5分钟,再瞬时降到25%溶液B重平衡10min。
图5为基因1011、4917失活的突变株与野生型菌株XM201的前体物定量结果。结果表明当琥珀酸脱氢酶被失活之后,在发酵前期胞内游离的前体物丙二酰辅酶A的水平升高,甲基丙二酰辅酶A的水平降低,乙酰辅酶A的水平未有明显变化。
步骤五,利用HPLC检测格尔登素的发酵产量
使用Agilent公司的Agilent 1200系列HPLC进行色谱分析,采用DAD紫外检测器测定304nm下的色谱吸收峰。色谱柱的参数为:Agilent Eclipse Plus C18,4.6×250mm,5μm;流动相流速为0.5mL/min;流动相:100%甲醇。柱温:室温。
图6-7为基因ORF3547、ORF1011、ORF6607和ORF4917敲除后格尔登素发酵水平检测。结果表明这些基因缺失之后,格尔登素的发酵水平有明显的提高,和出发菌株相比格尔登素的发酵产量提高约40%~100%,终产量达到1.3g/L。
- 还没有人留言评论。精彩留言会获得点赞!
- 用CRISPR/Cas9技术易于筛选获得目的基因敲除细胞系的方法及产品与制造工艺
- 一种Candida amazonensis的FLP/FRT基因敲除方法与制造工艺
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 利用CRISPR/Cas9敲除人PD‑1基因构建靶向CD19CAR‑T细胞的方法与制造工艺
- 一种构建基因替代小鼠研究野生型和突变体非肌性肌球蛋白重链II功能的方法与制造工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 猪ApoE基因敲除打靶载体及其构建方法和应用与制造工艺
- 一种血小板减少症的斑马鱼模型的制造方法与工艺
- 基于CRISPR技术敲除FUT8基因的方法与制造工艺