恶二唑类化合物的制备和应用的制作方法
本发明的实施例涉及合成领域,更具体地,涉及恶二唑类化合物的制备和应用。
背景技术:
白血病是一类造血干细胞恶性克隆性疾病。白血病按起病的缓急可分为急、慢性白血病。急性白血病细胞分化停滞在早期阶段,以原始及早幼细胞为主,疾病发展迅速,病程数月。慢性白血病细胞分化较好,以幼稚或成熟细胞为主,发展缓慢,病程数年。
慢性粒细胞白血病(chronic myelogenous leukemia,CML)是一种较为少见的起源于造血干细胞异常的恶性肿瘤,占所有癌症的0.3%左右。其主要特点是产生大量不成熟的白细胞,这些白细胞能够在骨髓内聚集,从而抑制骨髓的正常造血功能;并且其能够通过血液扩散到全身,因而导致病人出现贫血、血小板增多、脾大、感染、容易出血及气管感染等症状。慢性粒细胞白血病的发病率为1~2/10万,占所有白血病的18%,其发病人群在50岁以上较为常见,平均发病人群在65岁,男性和女性比例为1.4:1。在临床上根据人体骨髓中白细胞的数量和症状的严重程度,将CML分为三个期:慢性期(CP)、加速期(AP)和急变期(BP)。其中,90%的患者在早期诊断时处于慢性期,1-2年后约3%-4%的患者由慢性期进展为加速期,且3-6月后由加速期发展为急变期,如果患者没有接受任何治疗,其自然病程一般为3年左右。另外,据临床试验发现,约90%-95%的患者体内均可检测到费城染色体的存在,它是人类认识的第一个与恶性肿瘤相关的染色体异常改变,该染色体t(9;22)(q34;q11)是由9号染色体q34的c-Abl基因与22号染色体q11的c-Bcr基因相互易位而形成,其可以编码出慢性粒细胞的致病基因:BCR-ABL融合基因。该融合基因可以编码产生 具有高酪氨酸活性的BCR-ABL融合蛋白,与正常的c-ABL蛋白相比,该融合蛋白会不断地刺激白细胞增殖和介导细胞程序性死亡的耐受性而形成CML。由于CML对人类生命和健康的严重威胁,因此,针对如何治愈CML,控制血液和遗传学异常,消除症状,最大限度地延长生存期,依然是目前医学和药学领域内迫切需要解决的问题。
近年来,对于CML的治疗手段和策略都有了较大的进展,主要包括以下几种方法:第一种:化学药物治疗或联合化学治疗,常用药物如:羟基脲、马利兰、高三尖杉酯碱、阿糖胞苷等。根据临床结果显示,虽然在一定程度上这些药物可以改善症状,缓解病情,但是往往副反应较多,且不能明显延长病程或控制病情进展。第二种:干扰素α(INF-α)治疗CML。INF-α是一种生物糖蛋白,研究表明其具有抑制白细胞克隆增值,重塑染色体,调节免疫,抗肿瘤,抗病毒等功效。并曾在异基因干细胞移植无效时,作为治疗费城染色体阳性白血病的一线治疗药物,但是干扰素的治疗也存在较多的副反应,部分患者在使用干扰素4.2-20.4月内易产生干扰素抗体,并且停药后有复发的报告。第三种:造血干细胞移植包括两种:自体造血干细胞移植和异基因造血干细胞移植。处于慢性期的CML患者通过自体造血干细胞移植,可获得5年以上生存率高达40%。但是存在的主要缺陷是患者进行移植后细胞遗传学反应率低,不能从根本上消除白细胞恶性克隆。异基因造血干细胞移植是目前已证实唯一可以治愈费城染色体阳性白血病的手段。但由于合格骨髓提供者来源困难,大部分患者无法得到及时的治疗。另外,一部分患者在移植后存在抗宿主病、易感染、易复发及其他相关并发症等,因此选择移植对象时要尤为慎重。第四种:基因靶向治疗药物,即酪氨酸激酶抑制剂(TKI)。其主要是根据肿瘤细胞活动原理而设计的靶向抗癌药物,在治疗慢性粒细胞白血病中取得了较大的成功,给CML治疗带来了革命性的突破。
蛋白酪氨酸激酶(PTK)是细胞信号传导过程中的重要因子,它能够催化多种底物蛋白的磷酸化,从而传递信号,促进细胞生长、分化、增殖。BCR-ABL融合蛋白是一种高酪氨酸活性的蛋白激酶,它能不断催化ATP上γ-磷酸转移到蛋白酪氨酸残基上,激活下游信号通路,促使成熟CML 粒细胞无限增值,抑制细胞凋亡,继而造成造血干细胞的功能紊乱。由于BCR-ABL在正常细胞中不表达,因而抑制其活性成为CML治疗的一个新途径。
目前,临床使用的酪氨酸激酶抑制剂主要包括替尼类药物第一代(TKIs)伊马替尼,第二代(TKIs)尼罗替尼、达沙替尼和第三代(TKIs)ponatinib(AP245234)等。伊马替尼是由瑞士的诺华公司研制的第一个根据肿瘤产生机制而设计的靶向抗癌药物。其能够特异性的和BCR-ABL蛋白结合,从而阻断其下游信号通路,抑制费城染色体阳性细胞的复制与增值。另外,BCR-ABL融合基因在正常细胞中不表达,所以伊马替尼对正常造血细胞几乎无抑制作用。但是部分患者长时间服用后,均产生了药物的耐药性,继而导致病情复发。临床研究表明,酪氨酸激酶靶点的基因突变,导致药物分子无法与靶点结合,是药物耐药性的根本原因。为了克服BCR-ABL激酶突变,制药企业相继又研发出了两个新的酪氨酸激酶抑制剂,即:第二代药物尼罗替尼和达沙替尼。尼罗替尼是甲磺酸伊马替尼的类似物,其对野生型BCR-ABL蛋白的抑制活性较伊马替尼强20倍,并能有效克服对伊马替尼耐药的大部分BCR-ABL细胞突变株。达沙替尼是BCR-ABL激酶和Src族激酶的双重抑制剂,其体外抑制活性较甲磺酸伊马替尼高325倍。虽然达沙替尼和尼罗替尼能够克服大部分的BCR-ABL激酶突变,但是不能克服最重要的T315I突变,其虽仅占所有相关酪氨酸激酶突变的15%-20%,但却对以上所有酪氨酸激酶抑制剂治疗均无响应。
为了克服这一迫在眉睫的问题,很多药物研究单位都将研发出耐T315I突变的酪氨酸激酶抑制剂作为重点开发的项目。随后,大量的文献和综述报道了在这一领域取得的重要突破。例如:PPY-A,TG101113,BCR-ABL/Aurora双重激酶抑制剂陶扎色替(tozasertib),达努赛替(danusertib)等。其中tozasertib已经用于治疗耐药CML患者的临床试验中,但是缺陷是临床的给药必需采用静脉注射的方式。另外,由于较强的毒副作用,在临床治疗中其通常作为挽救疗法。尽管已取得这些进步,目前依然没有能够被批准治疗发生BCR-ABLT315I突变的药物。第三代酪氨酸激酶抑制剂ponatinib是一个新型的口服多靶点激酶抑制剂,除了能够抑 制未发生突变的酪氨酸激酶和目前所有类型突变的激酶,还能有效抑制T315I突变的BCR-ABL。因此,该药于2012年12月14日由FDA提前三个月批准上市。值得一提的是,由于ponatinib在后期患者治疗中发现存在“危及生命的血栓和血管重度狭窄”风险,已于2013年10月31号,被FDA要求暂停这种白血病药物的销售和推广。因此,研发出安全有效多靶点的抗耐药性的酪氨酸激酶抑制剂具有重要的价值和研究前景。
技术实现要素:
本发明提供了一类可用作酪氨酸激酶抑制剂的化合物,并且提供了这些化合物的合成方法。此外,通过实验验证了这些化合物的抑制酪氨酸激酶的活性。
本发明提供了一种化合物的制备方法,包括:在容器中加入第一化合物和第二化合物,在K2CO3和THF的作用下反应得到第三化合物,所述第一化合物为所述第二化合物为所述第三化合物为 其中,R为、所述第三化合物在N2H4-H2O和乙醇的作用下反应得到第四化合物,所述第四化合物为 所述第四化合物在THF和三乙胺的作用下与第五化合物反应得到第六化合物,所述第五化合物为所述第六化合物为 所述第六化合物在苯磺酰氯、碳酸钾和二氯甲烷的作用下反应得到第七化合物,所述第七化合物为所述第七化合物在CuI和Pd(PPh3)2Cl2的作用下与第八化合物反应得到第九化合物,所述第八化合物为所述第九化合物为第一目标化合物
在上述制备方法中,其中,当R为-CH2CH2OTBDPS时,还包括:所述所述第九化合物在乙酸、TBAF和THF的作用下反应得到第十化合物,所述第十化合物为第二目标化合物
在上述制备方法中,其中,当R为-CH2CH2NHBoc时,还包括:所述所述第九化合物在苯甲醚、二氯甲烷和三氟乙酸的作用下反应得到第十一化合物,所述第十一化合物为第三目标化合物
在上述制备方法中,还包括:所述第十一化合物在DCC、CH2Cl2和CS2的作用下反应得到第十二化合物,所述第十二化合物为第四目标化合物
在上述制备方法中,其中,所述第五化合物的合成方法包括:第十三化合物在SOCl2的作用下反应得到所述第五化合物,所述第十三化合物为
本发明还提供了根据以上制备方法得到的目标化合物在抑制酪氨酸激酶方面的用途。
本发明还提供了根据以上制备方法得到的目标化合物在治疗白血病中的用途。
附图说明
图1示出了化合物10a的1H NMR。
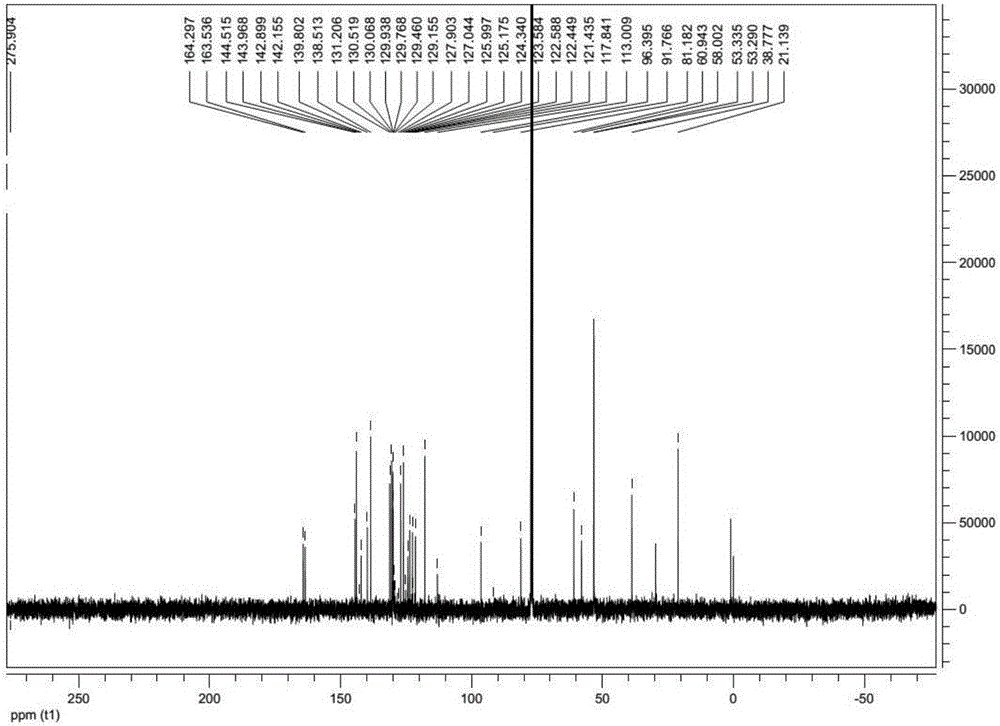
图2示出了化合物10a的13C NMR。
图3示出了化合物10d的1H NMR。
图4示出了化合物10d的13C NMR。
图5示出了化合物10e的1H NMR。
图6示出了化合物10e的13C NMR。
图7示出了化合物10f的1H NMR。
图8示出了化合物10f的13C NMR。
图9示出了阳性药ponatinib对k562细胞的活性。
图10示出了实例1-4(化合物10a、10d、10e、10f)对k562细胞的活性。
图11示出了ponatinib对HL60细胞的活性。
图12示出了实例1-4(化合物10a、10d、10e、10f)对HL60细胞的活性。
图13示出了ponatinib对KG1a细胞的活性。
图14示出了实例1-4(化合物10a、10d、10e、10f)对KG1a细胞的活性。
说明书中的英文简写对应的中文名称:
THF:四氢呋喃
TLC:薄层色谱(点板)
Pd(PPh3)Cl2:二氯化三苯基膦合钯(II)
DMF:二甲基甲酰胺
Et3N:三乙胺
TBAF:四丁基氟化铵三水化合物
DCC:N,N'-二环己基碳二亚胺
CH2CH2OTBDPS:醚类:叔丁基二苯基硅醚(TBDPS-OR)
叔丁基二苯基硅基(TBDPS)
Boc:叔丁氧羰基
具体实施方式
下面的实施例可以使本领域技术人员更全面地理解本发明,但不以任何方式限制本发明。
化合物的通式为:
其中,R为
该化合物的合成路线为:
1)具体合成步骤:
1.01化合物3a-3c的合成
在50mL的圆底烧瓶中,加入粗品化合物1(500mg,1.68mmol),化合物2(代表一类化合物)(3.36mmol),K2CO3(700mg,5.05mmol),20mL THF,室温搅拌4h,TLC监测反应进程。反应结束后,减压浓缩,用饱和NaHCO3溶液洗涤,二氯甲烷萃取,合并有机相,无水MgSO4干燥,过滤,浓缩,柱层析纯化,得到化合物3(实际上为化合物3a、3b或3c,为了简便,在无需对化合物3a、3b或3c区分的情况下可以称为化合物3,其他化合物也可以进行类似处理)。产率:80%-85%。
化合物3a:1H NMR(400MHz,CDCl3)δ8.26(s,1H),8.14(d,J=8Hz,1H),7.89(d,J=8Hz,1H),3.91(s,3H),3.68(s,2H),2.51(br,8H),2.30(s,3H)。
化合物3b:1H NMR(400MHz,CDCl3)δ8.30(s,1H),8.17(d,J=8.4Hz,1H),7.92(d,J=8.4Hz,1H),7.70-7.67(m,4H),7.42-7.36(m,6H),3.94(s,3H),3.81(t,J=6.4Hz,2H),3.69(s,2H),2.62-2.49(m,10H),1.05(s,9H)。
化合物3c:1H NMR(400MHz,CDCl3)δ8.27(s,1H),8.14(d,J=8Hz,1H),7.90(d,J=8Hz,1H),3.92(s,3H),3.68(br,2H),3.21(m,2H),2.48-2.44(m,10H),1.43(s,9H)。
1.02化合物4a-4c的合成
在25mL两口圆底烧瓶中加入化合物3(1.1mmol),N2H4-H2O(33mmol),6mL EtOH,70℃加热回流4h,TLC检测。反应结束后,减压浓缩,用饱 和NaHCO3溶液洗涤,二氯甲烷萃取,合并有机相,无水MgSO4干燥,过滤,浓缩,柱层析纯化,得到化合物4,产率80%-89%。
化合物4a:1H NMR(400MHz,CD3OD)δ8.13(s,1H),8.02(d,J=8.4Hz,1H),7.93(d,J=8.4Hz,1H),3.72(s,2H),2.53(br,8H),2.31(s,3H)。
化合物4b:1H NMR(400MHz,CDCl3)δ8.03(s,1H),7.93-7.87(m,2H),7.68-7.66(m,4H),7.59(s,1H),7.44-7.35(m,6H),3.82(t,J=6Hz,2H),3.68(s,2H),2.64-2.50(m,10H),1.04(s,9H)。
化合物4c:1H NMR(400MHz,CDCl3)δ8.05(s,1H),7.97(s,1H),7.90(d,J=2Hz,2H),3.67(s,2H),3.23-3.21(m,2H),2.49-2.47(m,10H),1.43(s,9H)。
1.03化合物7a-7c的合成
在50mL的圆底烧瓶中,化合物5(0.5g,1.91mmol),SOCl2(8mL)。将体系升温至70℃,回流搅拌3h后,反应物减压浓缩除去SOCl2,并用油泵抽干,得到化合物6即3-碘-4-甲基苯甲酰氯,为白色固体,直接投下一步。
在50mL的两口圆底烧瓶中,加入酰肼化合物4(1.6mmol),Et3N(3.2mmol),THF(15mL)。将体系冷却至0℃后,滴加化合物6的THF溶液(10mL)。滴毕,保持体系温度不变,继续搅拌2h,TLC监测反应进程,反应结束后,减压浓缩,用饱和NaHCO3溶液洗涤,二氯甲烷萃取,合并有机相,无水MgSO4干燥,过滤,浓缩,柱层析纯化,得到纯品化合物7为白色固体,产率65%-72%。
化合物7a:1H NMR(400MHz,CDCl3)δ8.29(d,J=1.2Hz,1H),8.12(s,1H),7.99(dd,J=1.2,8.4Hz,1H),7.91(d,J=8.4Hz,1H),7.73(dd,J=1.6,8Hz),7.29(d,J=8Hz,1H),3.69(s,2H),2.53-2.48(m,11H),2.32(s,3H)。
化合物7b:1H NMR(400MHz,CDCl3)δ8.25(d,J=1.2Hz,1H),8.12(s,1H),8.01(d,J=8.4Hz,1H),7.83(d,J=8.4Hz,1H),7.74(dd,J=1.2,8Hz,1H),7.68-7.66(m,4H),7.43-7.35(m,6H),7.17(d,J=8Hz,1H),3.80(t,J=6Hz,2H),3.63(s,2H),2.60-2.43(m,13H),1.04(s,9H)。
化合物7c:1H NMR(400MHz,CDCl3)δ8.26(d,J=1.6Hz,1H),8.09(s,1H),8.02(d,J=8Hz,1H)7.85(d,J=8.4Hz,1H),7.44(dd,J=1.6,7.6Hz,1H),7.02(d,J=8Hz,1H),3.65(s,2H),3.21(m,2H),2.51-2.44(m,13H),1.44(s,9H)。
1.04化合物8a-8c的合成
在50mL的圆底烧瓶中,依次加入化合物7(1.1mmol),TsCl(苯磺酰氯,1.65mmol),K2CO3(3.3mmol),CH2Cl2(15mL)。将混合物置于室温搅拌3h,TLC监测反应进程,反应结束后,用饱和NaHCO3溶液洗涤,二氯甲烷萃取,合并有机相,无水NaSO4干燥,过滤,浓缩,柱层析 纯化,得到纯品化合物8为白色固体,产率80%-86%。
化合物8a:1H NMR(400MHz,CDCl3)δ8.57(d,J=1.6Hz,1H),8.37(s,1H),8.29(d,J=8Hz,1H),8.05-8.01(m,2H),7.41(d,J=8Hz,1H),3.76(s,2H),2.59-2.53(m,11H),2.36(s,3H); 13C NMR(100MHz,CDCl3)δ163.56,163.35,145.87,142.14,136.99,131.18,130.18,129.93,129.64(J=31Hz),126.66,124.34(J=6Hz),123.77(J=272.8Hz),122.70,122.51,101.22,57.94,55.08,53.10,45.93,28.41。
化合物8b:1H NMR(400MHz,CDCl3)δ8.57(d,J=1.6Hz,1H),8.37(s,1H),8.29(d,J=8Hz,1H),8.04-8.01(m,2H),7.69-7.67(m,4H),7.44-7.36(m,7H),3.85(t,J=6Hz,2H),3.74(s,2H),2.69-2.53(m,13H),1.05(s,9H)。
化合物8c:1H NMR(400MHz,CDCl3)δ8.56(d,J=1.6Hz,1H),8.37(s,1H),8.28(d,J=8.4Hz,1H),8.02(dd,J=1.6,8Hz,1H),7.97(d,J=7.6Hz,1H),7.40(d,J=8Hz,1H),3.78(s,2H),3.37(br,2H),2.70-2.52(m,13H),1.44(s,9H)。
1.05化合物10a-10c的合成
在50mL两口圆底烧瓶中加入化合物9(0.2g,1.40mmol),1,3,4-噁二唑化合物8(1.27mmol)后,于手套箱加入催化剂CuI(21mg,0.11mmol),Pd(PPh3)2Cl2(39mg,0.06mmol)。将体系抽真空,冲Ar2,循环三次。用5mL注射器加入溶剂DMF(5mL),Et3N(0.3mL,2.01mmol)。将体系加热至65℃,搅拌10h。TLC检测反应进程,反应结束后,将混合物减压浓缩并用硅胶拌样后,通过硅胶柱分离纯化得到纯品化合物10为淡黄色固体。产率80%-89%。
化合物10a(实例1):1H NMR(400MHz,CDCl3)δ8.50(dd,J=1.2,4.4Hz,1H),8.38(s,1H),8.34(d,J=1.6Hz,1H),8.30(d,J=8Hz,1H),8.09(s,1H),8.07-8.00(m,3H),7.45(d,J=8Hz,1H),7.15(dd,J=4.4,9.2Hz,1H),3.74(s,2H),2.67(s,3H),2.56-2.51(m,8H),2.32(s,3H);13C NMR(100MHz,CDCl3)δ164.31,163.53,144.50,143.94,142.08,139.82,138.53,131.21,130.51,130.08,129.93,129.68(J=30.9Hz)127.04,125.99,124.36(J=5.8Hz),123.81(J=272.6Hz),123.61,122.67,121.46,117.80,113.03,96.40,81.20,57.97,55.16,53.15,46.00,21.12;HR MS(EI)calcd for C30H27F3N7O 558.2229[M+H]+;found 558.2217。
化合物10b:1H NMR(400MHz,CDCl3)δ8.51(dd,J=1.2,4.4Hz,1H),8.40(s,1H),8.34(d,J=1.6Hz,1H),8.31(d,J=8Hz,1H),8.09(s,1H),8.07-8.02(m,2H),7.96(d,J=8Hz,1H),7.66-7.64(m,4H),7.46-7.37(m,7H),7.16(dd,J=4.4,9.2Hz,1H),3.97(br,2H),3.78(s,2H),2.88-2.68(m,13H),1.05(s,9H)。
化合物10c:1H NMR(400MHz,CDCl3)δ8.51(dd,J=1.2,4.4Hz,1H),8.39(s,1H),8.35(d,J=1.6Hz,1H),8.30(d,J=8.4Hz,1H),8.09(s,1H),8.08-8.01(m,3H),7.46(d,J=8Hz,1H),7.16(dd,J=4.4,9.2Hz,1H),3.74(s,2H),3.23(m,2H),2.68(s,3H),2.53-2.47(m,10H),1.46(s,9H)。
1.06化合物10d(实例2)的合成
在50mL的圆底烧瓶中,加入化合物10b(0.5g,0.61mmol),CH3COOH (0.1mL,1.82mmol),TBAF(0.5mL,1.82mmol),THF(10mL)。将混合物于室温搅拌至化合物10b完全消失,反应结束后,减压浓缩除THF,用饱和NaHCO3溶液洗涤,二氯甲烷萃取,合并有机相,无水MgSO4干燥,过滤,浓缩,柱层析纯化,得到纯品化合物10d为淡黄色固体固体(0.23g,产率65%)。
1H NMR(400MHz,CDCl3)δ8.51(dd,J=1.2,4.4Hz,1H),8.39(s,1H),8.35(d,J=1.2Hz,1H),8.31(d,J=8Hz,1H),8.09(s,1H),8.07-8.01(m,3H),7.46(d,J=8Hz,1H),7.16(dd,J=4.4,9.2Hz,1H),3.75(s,2H),3.63(t,J=5.6Hz,2H),2.68(s,3H),2.60-2.58(m,10H);13C NMR(100MHz,CDCl3)δ164.32,163.51,144.52,143.95,141.98,139.81,138.52,131.21,130.52,130.07,129.94,129.68(J=31Hz),127.04,125.99,124.36(J=5.8Hz),123.81(J=272.6Hz),123.61,122.70,121.45,117.82,113.02,96.39,81.21,59.21,57.98,57.70,53.24,52.90,21.13;HR MS(EI)calcd for C31H29F3N7O2 588.2337[M+H]+;found588.1929.
1.07化合物10e(实例3)的合成
在50mL的两口圆底烧瓶中,加入化合物10c(0.73mmol),苯甲醚(anisole)(0.3mL),CH2Cl2(5mL),将体系冷却至0℃后,滴加CF3COOH(1mL)的CH2Cl2溶液(5mL)。滴毕,保持0℃搅拌1h后,体系移至室温搅拌,TLC监测反应进程,反应结束后,加入饱和Na2CO3溶液,调节体系PH至碱性。加入二氯甲烷萃取,合并有机相,无水Na2SO4干燥,减压浓缩并用硅胶拌样后,通过硅胶柱分离纯化(二氯甲烷:甲醇=20:1,氨水:0.5%)得到纯品化合物10e为淡黄色固体。
1H NMR(400MHz,CD3OD)δ8.63(dd,J=0.8,4Hz,1H),8.34(s,1H),8.30(d,J=8Hz,1H),8.20(s,1H),8.09-8.05(m,3H),7.99(dd,J=1.6,8Hz,1H),7.49(d,J=8Hz,1H),7.36(dd,J=4.4,9.2Hz,1H),3.75(s,2H),2.94(t,J=6.4Hz,2H),2.62-2.56(m,13H)13C NMR(100MHz,CDCl3)δ164.30,163.54,144.52,143.97,142.16,139.80,138.51,131.21,130.52,130.07,129.94,129.61(J=30.8Hz),127.04,126.00,124.34,123.81(J=272.6Hz),123.58,122.59,121.44,117.84,113.01,96.40,81.18,60.94,58.00,53.34,53.29,38.78,21.14;HR MS(EI)calcd for C31H30F3N8O 587.2495[M+H]+;found 587.2482。
1.08化合物10f(实例4)的合成
在50mL两口圆底烧瓶中加入化合物10e(0.51mmol),DCC(0.20g,0.77mmol),CH2Cl2(5mL),将体系冷却至0℃后,滴加CS2(0.31mL,5.1mmol)的CH2Cl2(5mL)溶液。滴毕,反应液保持0℃继续搅拌2h,TLC监测反应,反应结束后减压浓缩并用硅胶拌样后,通过硅胶柱分离纯化(二氯甲烷:甲醇=50:1)得到纯品化合物10f为淡黄色固体。
1H NMR(400MHz,CDCl3)δ8.50(d,J=3.6Hz,1H),8.37(s,1H),8.33(s,1H),8.29(d,J=8Hz,1H),8.08(s,1H),8.05-8.00(m,3H),7.44(d,J=8Hz,1H),7.15(dd,J=4.4,9.2Hz,1H),3.73(s,2H),3.59(t,J=6Hz,2H),2.71-2.57(m,13H);13C NMR(100MHz,CDCl3)δ164.30,163.49,144.50,143.95,141.95,139.81,138.52,132.61,131.22,130.51,130.04,129.93,129.67(J=31Hz),127.02,125.98,124.34,123.79(J=272.9Hz),123.60,122.70,121.43,117.82,113.01,96.39,81.22,57.92,57.20,53.13,52.95,43.07,21.12;HR MS(EI)calcd for C32H28F3N8OS 629.2059[M+H]+;found 629.2054。
活性测试
MTT粉末购买于Sigma公司,用磷酸缓冲液(PBS)将其配制成浓度为5毫克/毫升的溶液,选取0.22μm滤膜进行过滤,然后于4℃下避光保存。K562细胞(慢性粒细胞白血病细胞系)购于自北京金紫晶生物科技有限公司。实验中所使用的细胞培养基为改良型RPMI-1640(Hyclone)基础培养基,添加10%的胎牛血清(Hyclone)。
MTT比色法测定细胞活性包括下面几个步骤(以K562细胞的测试方法为例,KG1a HL60细胞的测试方法与K562细胞的方法一致):
(1)选取处于对数生长期的K562细胞,按每孔3×103接种于96孔板,5%CO2,37℃孵育培养过夜。
(2)加药,本实验中设置了9个浓度梯度,根据需要采用不同的浓度梯度,例如,体系中药物终浓度梯度为:10μM,3.33μM,1.11μM,0.37μM,0.12μM,0.04μM,0.013μM,0.004μM,0.0013μM。每个浓度5个复孔,同时设置对照组(不加药仅接种细胞)及空白孔(未接种细胞仅加培养基),5%CO2,37℃培养箱孵育72小时。
(3)每孔再加入20μL MTT溶液(5mg/ml,即0.5%MTT),继续培养4小时。若药物与MTT能够反应,可先离心后弃去培养液,小心用PBS冲洗2-3遍后,再加入含MTT的培养液。
(4)4小时后终止培养,小心地吸去孔内的液体。并向每孔加入150μL的二甲基亚砜。然后置于摇床上低速振荡15min左右,使结晶物充分溶解。采用酶联免疫检测仪MULTISKAN FC(Thermo scientific)测定490nm处及570nm各孔的吸光度值,测量时以空白孔作为调零孔。
(5)处理数据。以药物浓度为横坐标,细胞数为纵坐标,用数据处理软件SPSS软件(IBM公司)进行几率单位加权回归法(Bliss法)进行数据处理,作图,得到IC50值。见表1。
表1实例1-4化合物对K562、HL60、KG1a细胞的IC50值
由以上实例1-4对应的化合物的IC50的数据可知,合成的化合物对K562、KG1a、HL60细胞具有抑制活性,具有用于治疗白血病的前景。
本领域技术人员应理解,以上实施例仅是示例性实施例,在不背离本发明的精神和范围的情况下,可以进行多种变化、替换以及改变。
- 还没有人留言评论。精彩留言会获得点赞!