一种去甲阿奇霉素的制备方法与流程
本发明涉及大环内酯类抗生素制备
技术领域:
,尤其是一种阿奇霉素前体去甲阿奇霉素的制备方法。
背景技术:
:阿奇霉素是大环内酯类抗生素红霉素的结构改造物,广谱抗菌,对某些耐受β-内酰胺类抗生素的致病菌有效,具有组织渗透性强、浓度高,半衰期长的特点。临床上广泛用于治疗敏感致病菌感染引起的呼吸道、泌尿系统、皮肤软组织疾病,也可以用于性病治疗,防治性病传播。去甲阿奇霉素是制备阿奇霉素的前体,其经甲基化,得到阿奇霉素。阿奇霉素的制备过程包括:硫氰酸红霉素a(ii)与羟胺发生肟化反应,得到硫氰酸红霉素a肟(iii),经磺酰氯处理重排,得到红霉素a亚胺醚{红霉素a6,9-亚胺醚(iv)和异构体红霉素a11,9-亚胺醚(v)},经过催化氢化或用化学试剂硼氢化钾、硼氢化钠还原,得到去甲阿奇霉素(i),用甲酸、甲醛甲基化得到阿奇霉素。阿奇霉素的制备过程反应方程式如下:在阿奇霉素的制备过程中,肟化、重排和甲基化反应过程研究较为充分,工艺条件简单,收率高,但亚胺醚还原制备去甲阿奇霉素过程复杂,是影响阿奇霉素收率和质量的关键步骤。文献报道亚胺醚催化氢化还原制备去甲阿奇霉素如下:us:5,586,587(1997年)报道,亚胺醚在乙酸中,pto2为催化剂,50atm室温催化氢化48h,得到去甲阿奇霉素,收率85.8%。cn:1199736a(1998年)报道了亚胺醚催化氢化和甲基化“一锅法”,制备阿奇霉素的方法,乙酸为溶剂,5%rh/c为催化剂,70atm,40℃氢化3h,加入过量甲醛溶液,40atm,40℃继续反应20h,直接制备阿奇霉素。us:5,869,629(1999年)报道6,9-亚胺醚(iv)醋酸水溶液,在75atm下用5%pa/c为催化剂氢化12h,得到去甲阿奇霉素。us:6,528,492b1(2003年)报道以红霉素a为起始原料,肟化、重排、还原、甲基化“一锅法”制备阿奇霉素的方法;其中用ni-al为催化剂,85atm氢化还原亚胺醚制备去甲阿奇霉素。us:2005/0222052a1公开了亚胺醚磷酸水溶液,以5%pt/c为催化剂,在15℃、20atm下催化氢化24h,得到去甲阿奇霉素。cn:103087125a公开了亚胺醚甲醇溶液,用高氯酸调节ph=5-6,在pt/c催化下,于40--43℃、8.3-8.6atm下氢化还原4h,制备去甲阿奇霉素。化学试剂法还原亚胺醚制备去甲阿奇霉素,涉及到去甲阿奇霉素硼酸酯和双去甲阿奇霉素硼酸酯2个中间产物。酸解脱除硼酸酯,在碱性中和分离过程中部分发生可逆反应,重新转化成去甲阿奇霉素硼酸酯,造成收率低、分离困难。采取酸解脱除硼酸酯时加入多羟基有机助剂,快速加碱中和,可以部分抑制可逆反应,提高收率。wo:03/082889a1公开了6,9-亚胺醚在甲醇溶剂中,用6倍摩尔数的硼氢化钠低温还原制备去甲阿奇霉素,酸解过程加入柠檬酸,收率85.5%。us:6,703,372b1(2004年)公开了用7.7倍摩尔数的硼氢化钠还原,得到双去甲阿奇霉素硼酸酯,在多羟基树脂存在下酸解,制备去甲阿奇霉素。us:6,451,990b1(2002年)公开了6,9-亚胺醚在甲醇溶剂中,用硼氢化钠还原,分离得到去甲阿奇霉素硼酸酯,用甲醛、甲酸甲基化转化成阿奇霉素硼酸酯,酸解,用氢氧化钙溶液中和,分离得到阿奇霉素,收率低于50%。马敏等在《精细化工》vol.23,no.8中报道了用硼氢化钾还原亚胺醚,酸解、中和萃取,直接进行甲基化后,再进行酸解,制备阿奇霉素。cn:101712703a(2010年)和cn:102127064a(2011年)公开的制备方法类似,6,9-亚胺醚在ph=5-6水溶液中,用硼氢化钾还原,在多羟基助剂存在下水解,加碱中和,二氯甲烷萃取,得到去甲阿奇霉素和去甲阿奇霉素硼酸酯混合溶液,进行甲基化,再进行二次酸解、中和,制备阿奇霉素。采用催化氢化技术将亚胺醚转化成去甲阿奇霉素的优势在于还原过程没有去甲阿奇霉素硼酸酯中间产物生成。不足之处设备投资大,使用贵金属催化剂,反应物处理不好,易出现催化剂中毒,造成生产成本高。另外,实验过程中,本课题组发现,去甲阿奇霉素在酸性水、甲醇、乙腈中不稳定,室温的条件下,出现杂质,纯度下降;因此,亚胺醚在弱酸性水或醋酸中催化加氢制备去甲阿奇霉素过程中,不可避免出现副反应,出现收率低,影响最终阿奇霉素产品质量。使用化学试剂还原亚胺醚制备去甲阿奇霉素,常压进行反应,设备投资小;但由于酸解、中和过程存在部分可逆反应,生成去甲阿奇霉素硼酸酯和双去甲阿奇霉素硼酸酯2个中间产物,造成分离步骤繁琐,需要进行多次有机溶剂萃取,生产效率低下。另外文献公开的硼氢化钠为还原剂,需要甲醇为溶剂,低温操作,硼氢化钠用量大,生产成本高。针对现有技术的不足,需要开发操作简单、收率高的亚胺醚制备去甲阿奇霉素的方法,解决阿奇霉素生产过程的关键技术问题。技术实现要素:本发明要解决的技术问题是提供一种去甲阿奇霉素的制备方法,该方法提高了还原剂的利用率,可以控制硼酸酯的逆反应,操作简单,减少不必要的分离过程,得到高质量去甲阿奇霉素。为解决上述技术问题,本发明所采取的技术方案是:一种去甲阿奇霉素的制备方法,包括如下步骤:(1)还原:红霉素a6,9-亚胺醚(iv)在水中,0℃-室温,ph=7.0-9.0下,用还原试剂硼氢化钠或硼氢化钾还原;(2)水解:之后在有机溶剂和水存在下,0℃-室温,ph=2.0-3.5下,进行连续水解;再将连续水解两相反应液直接加入碱性水溶液中,调节ph≥12,搅拌,分层,弃去水相;其中,有机溶剂为二氯甲烷、氯仿、1,2-二氯甲烷、乙酸乙酯或乙酸丁酯;(3)连续水解的最后一次水解反应结束后,分层,水相直接加入碱性水溶液中,调节ph≥12,搅拌,析出去甲阿奇霉素(i),反应方程式如下:优选的,步骤(1)中,反应温度为0-5℃,还原试剂为硼氢化钾。优选的,步骤(1)中,红霉素a6,9-亚胺醚(iv)是由红霉素a或硫氰酸红霉素a(ii)经肟化、重排得到的。优选的,步骤(1)中,硼氢化钠或硼氢化钾摩尔数为红霉素a6,9-亚胺醚(iv)摩尔数的1.2-1.5倍。优选的,步骤(2)中,有机溶剂为二氯甲烷或氯仿。优选的,步骤(2)中,反应温度为0-5℃,ph=2.5-3.0下,进行连续水解。优选的,步骤(2)中,连续水解2-3次,每次水解至水解原料≤1wt%。优选的,步骤(2)中,连续水解调节ph=2.0-3.5所用酸为盐酸、硫酸、磷酸;步骤(2)和(3)中,碱性水溶液为氢氧化钠水溶液、氢氧化钾水溶液、氢氧化钙水溶液、碳酸钠水溶液或氨水。进一步优选的,步骤(2)中,连续水解调节ph=2.0-3.5所用酸为10wt%-20wt%盐酸;步骤(2)和(3)中,碱性水溶液为氢氧化钠水溶液。优选的,去甲阿奇霉素的制备方法,包括如下步骤:(1)还原:将红霉素a6,9-亚胺醚(iv)悬浮于3-8倍质量的水中,搅拌,降温0-5℃,加入盐酸、硫酸或磷酸,调节ph=7-0-7.5,滴加5-10wt%的硼氢化钠或硼氢化钾水溶液,当ph=9.0时,停止滴加5-10wt%的硼氢化钠或硼氢化钾水溶液,继续搅拌反应1-2h,tlc监测反应,如有红霉素a6,9-亚胺醚(iv)原料点存在,再滴加盐酸、硫酸或磷酸,调节ph=7.0-7.5后,继续滴加5-10wt%的硼氢化钠或硼氢化钾水溶液,至反应完全;用酸中和至ph=6-7,搅拌至无气体放出为止;加入红霉素a6,9-亚胺醚(iv)质量数的5-15倍质量的有机溶剂二氯甲烷、氯仿、1,2-二氯甲烷、乙酸乙酯或乙酸丁酯,用10-20wt%氢氧化钠水溶液调节ph≥11,搅拌,静置分层;水相弃去;(2)水解:有机相中加入红霉素a6,9-亚胺醚(iv)质量数的3-6倍质量的水,搅拌,降温0-5℃,加入盐酸、硫酸或磷酸,调节ph=2.5-3.0,进行第1次水解,tlc监测至硼酸酯原料点消失;将水解两相反应液加入10-20wt%氢氧化钠水溶液中,调节ph≥12,搅拌,静置分层,弃去水相;有机相中继续加入红霉素a6,9-亚胺醚(iv)质量数的4-6倍质量的水,搅拌,降温0-5℃,加入盐酸、硫酸或磷酸,调节ph=2.5-3.0,进行第2次水解,tlc监测至硼酸酯原料点消失;将水解两相反应液加入到10-20wt%氢氧化钠水溶液,调节ph≥12,搅拌,静置分层,再次弃去水相;有机相中再加入红霉素a6,9-亚胺醚(iv)质量数的3-6倍质量的水,搅拌,降温0-5℃,加入盐酸、硫酸或磷酸,调节ph=2.5-3.0,进行第3次水解,tlc监测至硼酸酯原料点消失;(3)分层,有机相保留套用;20℃-30℃,水相加入到5%氢氧化钠水溶液中,保温搅拌,调节ph≥12,继续搅拌,析出去甲阿奇霉素(i),过滤,滤饼水洗,得到含量85wt%以上去甲阿奇霉素(i)粗品,直接进行甲基化反应,得阿奇霉素。本发明在化学试剂还原红霉素a亚胺醚制备去甲阿奇霉素的过程中,中性及弱碱性(ph=7.0-9)的条件下,硼氢化钠或硼氢化钾可以顺利还原红霉素a6,9-亚胺醚(iv),当ph≥8,得到双去甲阿奇霉素硼酸酯(vi)为主的反应物和部分去甲阿奇霉素硼酸酯(vii)。在中性及弱酸性(ph=4.0-7.0)条件下,双去甲阿奇霉素硼酸酯(vi)转化成去甲阿奇霉素硼酸酯(vii);继续增加酸度,去甲阿奇霉素硼酸酯(vii)水解去除硼酸,经过碱中和,析出去甲阿奇霉素(i)。反应方程式如下:在硼氢化钠或硼氢化钾还原完成后,加入碱中和析出去甲阿奇霉素过程中,本课题组意外发现双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)均易溶于碱性水中,而去甲阿奇霉素不溶于碱性水中,尤其是当ph≥12时,去甲阿奇霉素几乎不溶于水。通过将反应液加入碱水中析出去甲阿奇霉素(i)固体,能够减少逆反应硼酸酯的形成,少量双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)溶于碱性水,能够有效分离得到高质量去甲阿奇霉素(i)。通过采用中性及弱碱性的条件下,以水为溶剂,用硼氢化钠或硼氢化钾还原红霉素a6,9-亚胺醚(iv);反应完成后,加入有机溶剂萃取反应液,弃去含有过量的硼氢化钠或硼氢化钾的水相。有机相加入水,调至酸性水解,快速加入到碱溶液中和至ph≥12,静止分层,再次弃去水相。加入水,调节至酸性,进行二次水解。反应完成后,分相,将水相加入碱性水中,析出去甲阿奇霉素,经过滤,水洗,烘干,可以得到几乎不含硼酸酯的去甲阿奇霉素(i),去甲阿奇霉素(i)和其硼化物得到有效分离。滤液中少量去甲阿奇霉素硼酸酯(vii)经有机相萃取,可重复使用。本发明使用硼氢化钠或硼氢化钾还原红霉素a6,9-亚胺醚(iv)在ph=7-9范围内进行。与已知的硼氢化钠还原相比,减少了硼氢化钠用量,不需使用甲醇溶剂。与已知的硼氢化钾还原相比,减少ph=5-6时硼氢化钾分解,减慢氢气的放出速度,提高反应过程的安全性。在有机相和水相中通过两次水解、加入到碱液中和;第三次水解的水相加入碱水中析出产物,达到去甲阿奇霉素(i)与其硼酸酯的有效分离,得到高质量产物,可以直接进行甲基化,制备阿奇霉素。采用上述技术方案所产生的有益效果在于:(1)本发明的去甲阿奇霉素的制备过程,使用硼氢化钾或硼氢化钠为还原剂,在中性及弱碱性水相中进行反应,减少了还原剂的酸性无效分解,提高了还原剂的利用率,减慢氢气的放出速度,增加生产的安全性;(2)脱除硼酸酯的过程在有机相存在下酸性水解,加入到碱性水溶液中和,保证了加入过程始终在碱性条件下完成,析出的去甲阿奇霉素直接进入有机相,减少可逆反应的发生;(3)经过2次有机相存在下水解,第3次水解分层,水相直接加入到碱性水溶液中,析出去甲阿奇霉素固体,去甲阿奇霉素及其硼酸酯得到有效分离,得到高质量去甲阿奇霉素产物,操作简单,节约生产成本,有机相循环套用;得到的去甲阿奇霉素固体可以直接进行甲基化,生产高质量阿奇霉素。附图说明下面结合附图和具体实施方式对本发明作进一步详细的说明;图1是本发明参考例1-2中制得的红霉素a肟硫氰酸盐的高效液相色谱图;图2是本发明参考例2中制得的红霉素a亚胺醚的高效液相色谱图;图3是本发明实施例2中制得的去甲阿奇霉素的高效液相色谱图;图4是本发明参考例3-2中制得的阿奇霉素的高效液相色谱图。具体实施方式实施例只是为了具体说明本发明过程,并不是对
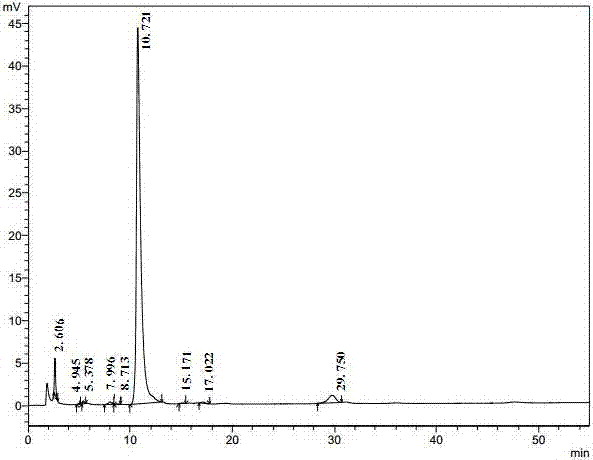
发明内容进行限制。参考例1-1:硫氰酸红霉素a肟(iii)的制备1000ml三口瓶加入盐酸羟胺(114g,1.64mol),甲醇300g,室温搅拌,滴加三乙胺调节ph=6.3-6.7。加入硫氰酸红霉素a(ii)(200g,0.25mol),升温至30-35℃反应4h,反应过程滴加三乙胺调节ph=6.5±0.2;升温至50-55℃,继续保温反应48h。降温至15-20℃,滴加600ml去离子水,搅拌0.5h;0-5℃析晶2h。过滤,用200ml去离子水洗涤,干燥,得白色固体红霉素a肟硫氰酸盐(iii)180g。收率88.4%,熔点182-185℃,纯度93.7%(hplc)。参考例1-2:硫氰酸红霉素a肟(iii)的制备2000ml三口瓶加入盐酸羟胺(228g,3.3mol),甲醇600g,室温搅拌,滴加三乙胺调节ph=6.3-6.7。加入硫氰酸红霉素a(ii)(400g,0.5mol),升温至30-35℃,保温反应10h;反应过程滴加三乙胺调节ph=6.5±0.2;升温至50-55℃,继续保温反应40h。降温至15-20℃,滴加1200ml去离子水,搅拌0.5h;0-5℃,析晶2h。过滤,用400ml去离子水洗涤,烘干,得白色固体红霉素a肟硫氰酸盐(iii)348g,收率87.0%,熔点182-184℃,纯度93.8%(hplc见图1和表1所示)。表1红霉素a肟硫氰酸盐的hplc检测结果峰#保留时间面积面积%高度分离度拖尾因子理论塔板#15.1894515.0210.3091960.0000.0001417.54725.9252320.6390.1591451.5050.0003081.12137.5965082.1960.3473474.1171.1096233.25049.06018683.7101.2778473.4361.4716011.604510.83238186.5962.60918343.6180.9607165.879612.99918449.4041.2615743.1411.0483588.149716.1971374109.69593.890409443.9331.2607307.045824.2962191.0190.1507410.6091.12715975.680总计1463538.280100.00044960表1中,保留时间16.197min对应的峰为红霉素a肟硫氰酸盐,12.99min处为硫氰酸红霉素a。参考例2:红霉素a亚胺醚的制备2000ml三口瓶中加入红霉素a肟硫氰酸盐(iii)(180g,0.22mol)、丙酮540ml,搅拌。0-10℃,加入10%碳酸氢钠溶液450g,5℃以下,滴加对甲苯磺酰氯(51g,0.27mol),1h滴毕,保温反应3h。反应液中滴加1200ml去离子水,用10wt%naoh水溶液调节ph=11-12,搅拌1h。0-5℃析晶1h。过滤,室温条件下用400ml去离子水洗涤,烘干得142g白色固体红霉素a亚胺醚。收率88.3%,熔点134-140℃,纯度92.9%(hplc见图2和表2所示)。表2红霉素a亚胺醚的hplc检测结果峰#保留时间面积面积%高度分离度拖尾因子理论塔板#12.60635405.1422.50842990.0001.4192051.63724.9451317.4080.0931319.0771.1014807.32735.3782546.6540.1802181.4271.0854458.05647.9967476.0170.5303115.6030.0002729.04058.7131087.8390.077571.2550.0004329.944610.7211311764.75392.913443233.3441.8304076.105715.1711777.7730.126726.8350.8399266.035817.0223963.5930.2811362.6891.6628324.875929.75046479.6873.29287912.1090.8337701.607总计1411818.866100.00050425表2中,保留时间10.721min对应的峰为红霉素a6,9-亚胺醚,经本发明的方法制得的红霉素a亚胺醚,经hplc检测9,11亚胺醚的含量均小于1%。实施例1:去甲阿奇霉素(i)制备1.1:还原40g红霉素a亚胺醚悬浮于160ml水中,搅拌10min,降温至0-5℃,滴加10wt%盐酸调节ph=7.0,缓慢加入12g16.7wt%硼氢化钾水溶液,1h加毕,保温搅拌反应2h,ph=8.5-9.0。滴加10wt%盐酸,调节ph=7.0,滴加6.2g19wt%硼氢化钾水溶液,保温搅拌反应2h,ph=8.0-8.5。滴加10wt%盐酸,调节ph=8.0,滴加6.2g19wt%硼氢化钾水溶液,保温反应3h,tlc监测,反应结束。滴加10wt%盐酸,调节ph=5-6,搅拌至无气体放出为止。加入280ml二氯甲烷,滴加10wt%氢氧化钠水溶液调节ph=11-12,搅拌0.5h,静止分层。水层用120ml二氯甲烷萃取。合并二氯甲烷层,水层弃去。1.2:一次水解有机相二氯甲烷层中加入160ml水,降温至0-5℃,滴加10wt%盐酸调节水相ph=2.5-3.0,保温搅拌反应1h。tlc监测,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}基本消失。搅拌下将反应液倾入100g5wt%的氢氧化钠水溶液中,室温搅拌1h,静止分层,水层弃去。1.3:二次水解上步有机相二氯甲烷层加入160ml水,降温至0-5℃,10%盐酸调ph=2.5-3.0,保温搅拌反应1h。tlc监测,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}消失。搅拌下将反应液倾入100g5wt%的氢氧化钠水溶液中,室温搅拌1h,静止分层,水层弃去。1.4:去甲阿奇霉素(i)的制备二次水解的二氯甲烷层加入160ml水,降温至0-5℃,10wt%盐酸调ph=2.5-3.0,保温搅拌反应1h。tlc监测硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}水解彻底,静止分层。将水层快速倒入200g2.5wt%氢氧化钠水溶液中,搅拌0.5h。25-30℃减压除去残存二氯甲烷,复测ph=12-13。20-25℃搅拌析晶1h,过滤,用40ml水洗涤2次,烘干,得去甲阿奇霉素(i)35.6g,收率89.0%,熔点126-128℃,纯度84.5%,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)之和}≤1%(hplc)。实施例2:去甲阿奇霉素制备2.1:还原80g红霉素a亚胺醚悬浮于320ml水中,搅拌10min,降温至0-5℃,滴加磷酸调节ph=7.0,缓慢加入16.7wt%硼氢化钾水溶液20g,1h加毕,保温搅拌反应2h,ph=8.5-9.0。滴加磷酸,调节ph=7.0,滴加20wt%硼氢化钾水溶液10g,保温搅拌反应2h,ph=8.0-8.5。滴加磷酸,调节ph=8.0,继续滴加20wt%硼氢化钾水溶液10g,保温反应3h,tlc监测,反应结束。滴加10wt%盐酸,调节ph=5-6,搅拌至无气体放出为止。加入800ml二氯甲烷,滴加10wt%氢氧化钠水溶液调节ph=11-12,搅拌0.5h,静止分层。水层用200ml二氯甲烷萃取。合并二氯甲烷层,水层弃去。将二氯甲烷层平均分为两份。2.2:一次水解步骤2.1的一份二氯甲烷层中加入160ml水,降温至0-5℃,滴加10wt%盐酸调节水相ph=2.5-3.0,保温搅拌反应1h。tlc监测,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}基本消失。搅拌下将反应液倾入5wt%的氢氧化钠水溶液100g中,室温搅拌1h,静止分层,水层弃去。2.3:二次水解上步有机相加入160ml水,降温至0-5℃,10wt%盐酸调ph=2.5-3.0,保温搅拌反应1h。tlc监测,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}消失。搅拌下将反应液倾入5wt%的氢氧化钠水溶液100g中,室温搅拌1h,静止分层,水层弃去。2.4:去甲阿奇霉素(i)的制备二次水解的二氯甲烷层加入160ml水,降温至0-5℃,10wt%盐酸调ph=2.5-3.0,保温搅拌反应1h。tlc监测硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}消失,静止分层。将水层快速倒入3wt%氢氧化钠水溶液200g中,搅拌0.5h。25-30℃减压除去残存二氯甲烷,控制ph=12-13。20-25℃搅拌析晶1h,过滤,用50ml水洗涤2次,烘干,得去甲阿奇霉素(i)35.3g,收率88.3%,熔点127-129℃,纯度85%,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}未检出(hplc)。步骤2.1的另一份二氯甲烷操作同上,得去甲基阿奇霉素(i)35.4g,收率88.5%,熔点为127-129℃,纯度87.0%,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)之和}≤0.5%(hplc见图3和表3所示)。表3去甲基阿奇霉素的hplc检测结果峰#保留时间面积面积积%高度分离度拖尾因子理论塔板#12.33810128.8030.9848860.0001.706787.83624.4676406.6780.6233284.6200.000922.77534.9982576.7230.2501390.8740.0001015.12646.17679631.3477.73842952.2281.2463313.01957.66122343.5512.1719943.2370.7303930.765611.390896084.68187.076349566.8721.3745831.552715.6874347.9060.4231897.1960.92911021.376822.7784343.1990.42214010.1711.00513080.556930.3523222.7970.313708.1591.42413121.502总计1029085.684100.00041997表3中,保留时间11.390min对应的峰为去甲基阿奇霉素。实施例3:去甲阿奇霉素(i)制备3.1:还原80g红霉素a亚胺醚悬浮于320ml水中,搅拌10min,降温至0-5℃,滴加10wt%硫酸调节ph=7.0,缓慢加15wt%硼氢化钾水溶液20g,1h加毕,保温搅拌反应2h,ph=8.5-9.0。滴加10wt%硫酸,调节ph=7.0,滴加20wt%硼氢化钾水溶液10g,保温搅拌反应2h,ph=8.0-8.5。再次滴加10wt%盐酸,调节ph=8.0,继续滴加20wt%硼氢化钾水溶液11g,保温反应3h,tlc监测,反应结束。滴加10wt%硫酸,调节ph=5-6,搅拌至无气体放出为止。加入800ml二氯甲烷,滴加10wt%氢氧化钠水溶液调节ph=11-12,搅拌0.5h,静止分层。水层用200ml二氯甲烷萃取。合并二氯甲烷层,水层弃去。3.2:一次水解有机相二氯甲烷层中加入320ml水,降温至0-5℃,滴加10wt%硫酸调节水相ph=2.5-3.0,保温搅拌反应1h。tlc监测,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}基本消失。搅拌下将反应液倾入5wt%的氢氧化钠水溶液200g中,室温搅拌1h,静止分层,水层弃去。3.3:二次水解上步有机相加入320ml水,降温至0-5℃,用10wt%硫酸调ph=2.5-3.0,保温搅拌反应1h。tlc监测,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}消失。搅拌下将反应液倾入5wt%的氢氧化钠溶液200g中,室温搅拌1h,静止分层,水层弃去。3.4:去甲阿奇霉素(i)的制备二次水解的二氯甲烷层加入320ml水,降温至0-5℃,10wt%硫酸调ph=2.5-3.0,保温搅拌反应1h。tlc监测硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}水解彻底,静止分层。将水层快速倒入400g2.5wt%-3wt%氢氧化钠水溶液400g中,搅拌0.5h。25-30℃减压除去残存二氯甲烷,控制ph=12-13。20-25℃搅拌析晶1h,过滤,用120ml水洗涤2次,烘干,得去甲阿奇霉素(i)70.5g,收率88.0%,熔点128-130℃,纯度80%,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)之和}≤0.3%(hplc)。实施例4:去甲阿奇霉素(i)的制备4.1:还原80g红霉素a亚胺醚悬浮于320ml水中,搅拌10min,降温至0-5℃,滴加50wt%醋酸调节ph=7.0,缓慢加15wt%硼氢化钾水溶液20g,1h加毕,保温搅拌反应2h,ph=8.5-9.0。滴加50wt%醋酸,调节ph=7.0,滴加20wt%硼氢化钾水溶液10g,保温搅拌反应2h,ph=8.0-8.5。再次滴加50wt%醋酸,调节ph=8.0,继续滴加20wt%硼氢化钾水溶液11g,保温反应3h,tlc监测,反应结束。滴加50wt%醋酸,调节ph=5-6,搅拌至无气体放出为止。加入800ml二氯甲烷,滴加10wt%氢氧化钠水溶液调节ph=11-12,搅拌0.5h,静止分层。水层用200ml二氯甲烷萃取。合并二氯甲烷层,水层弃去。4.2:一次水解有机相二氯甲烷层中加入320ml水,降温至0-5℃,滴加85wt%磷酸调节水相ph=2.5-3.0,保温搅拌反应1h。tlc监测,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}基本消失。搅拌下将反应液倾入5wt%的氢氧化钠溶液200g中,室温搅拌1h,静止分层,水层弃去。4.3:二次水解上步有机相二氯甲烷层加入320ml水,降温至0-5℃,用85wt%磷酸调ph=2.5-3.0,保温搅拌反应1h。tlc监测,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}消失。搅拌下将反应液倾入5wt%的氢氧化钠水溶液200g中,室温搅拌1h,静止分层,水层弃去。4.4:去甲阿奇霉素(i)的制备二次水解的二氯甲烷层加入320ml水,降温至0-5℃,85wt%磷酸调ph=2.5-3.0,保温搅拌反应1h。tlc监测硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)}水解彻底,静止分层。将水层快速倒入400g2.5wt%-3wt%氢氧化钠水溶液400g中,搅拌0.5h。25-30℃减压除去残存二氯甲烷,控制ph=12-13。20-25℃搅拌析晶1h,过滤,用120ml水洗涤2次,烘干,得去甲阿奇霉素(i)72.0g,收率90.0%,熔点128-130℃,纯度84%,硼酸酯{双去甲阿奇霉素硼酸酯(vi)和去甲阿奇霉素硼酸酯(vii)之和}≤0.3%(hplc)。参考例3-1:阿奇霉素的制备250ml三口瓶加入去甲基阿奇霉素(i)(15g,0.02mol),丙酮50ml,室温搅拌溶解。30-40℃,滴加无水甲酸(2.5g,0.06mol),37%甲醛(2.2g,0.03mol)。升温至50-55℃,保温反应4h,tlc监测反应完毕。降至室温,加入50ml去离子水,10wt%氢氧化钠水溶液调节ph=12-13,滴加50ml去离子水,搅拌析晶2h。过滤,用30ml水洗涤2次,烘干,得阿奇霉素13.5g,收率90%,纯度97.6%(hplc)。参考例3-2:阿奇霉素的制备500ml三口瓶加入去甲基阿奇霉素(i)(50g,0.07mol),丙酮175ml,室温搅拌溶解。30-40℃,滴加无水甲酸(8.5g,0.18mol),37%甲醛(7.3g,0.09mol)。升温至50-55℃,保温反应4h,tlc监测反应完毕。降至室温,加入175ml去离子水,10wt%氢氧化钠水溶液调节ph=12-13,滴加175ml去离子水,搅拌析晶2h。过滤,用100ml水洗涤2次,烘干,得阿奇霉素47.5g,收率95%,纯度98.8%(hplc见图4和表4所示)。表4阿奇霉素的hplc检测结果保留时间面积面积%高度分离度(usp)拖尾因子理论塔板数(usp)23.588240460.065714--1.1001067525.247833410.22510601.2181.442311128.800709280.19214532.3001.113810436.294187340.0514074.044--358137.285252980.0685450.511--1053541.623124220.0342763.1631.2361665049.1143656101598.8943099193.3542.542382760.578319330.0864754.475--1546562.410479330.1308221.0380.9392488364.922944030.25514441.5121.0652229436970055100.000317114表4中,保留时间49.114min对应的峰为阿奇霉素。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1