作为ALK和FAK双重抑制剂的稠合双环2,4‑二氨基嘧啶衍生物的制作方法
技术领域
本发明涉及可用于治疗ALK或FAK介导的障碍或病症的ALK和/或FAK抑制剂。
背景技术:
间变性淋巴瘤激酶(ALK)是一种跨细胞膜受体酪氨酸激酶,其属于胰岛素受体亚族。ALK的最丰富的表达发生在新生儿脑中,表明ALK在脑发育中可能起作用(Duyster,J.等,Oncogene,2001,20,5623-5637)。
ALK也与某些肿瘤的发展有关。例如,约60%的间变性大细胞淋巴瘤(ALCL)伴有产生由核磷蛋白(NPM)和ALK的细胞内结构域构成的融合蛋白的染色体突变。(Armitage,J.O.等,《癌症:肿瘤学原理与实践》(Cancer:Principle and Practice of Oncology),第6版,2001,2256-2316;Kutok J.L.&Aster J.C.,J.Clin.Oncol.,2002,20,3691-3702)。这种突变蛋白NPM-ALK具有组成性活性的酪氨酸激酶结构域,其通过下游效应子的激活而负责其致癌性质。(Falini,B.等,Blood,1999,94,3509-3515;Morris,S.W.等,Brit.J.Haematol.,2001,113,275-295;Duyster等,Kutok&Aster)。此外,转化的EML4–ALK融合基因已在非小细胞肺癌(NSCLC)患者中被鉴定到(Soda,M.等,Nature,2007,448,561-566),并且代表了作为用于ALK抑制剂疗法的有希望的靶点的ALK融合蛋白名单中的另一个成员。实验数据已证实,组成性活性的ALK的异常表达与ALCL的病理发生直接相关,并且ALK的抑制可以明显削弱ALK+淋巴瘤细胞的生长(Kuefer,Mu等,Blood,1997,90,2901-2910;Bai,R.Y.等,Mol.Cell Biol.,1998,18,6951-6961;Bai,R.Y.等,Blood,2000,96,4319-4327;Ergin,M.等,Exp.Hematol.,2001,29,1082-1090;Slupianek,A.等,Cancer Res.,2001,61,2194-2199;Turturro,F.等,Clin.Cancer Res.,2002,8,240-245)。在约60%的炎性成肌纤维细胞瘤(IMT)、一种主要影响儿童和年轻成人的缓慢生长的肉瘤中,也已经证实了组成性激活的嵌合ALK。(Lawrence,B.等,Am.J.Pathol.,2000,157,377-384;Duyster等)。
此外,ALK及其推测的配体多效营养因子在人类成胶质细胞瘤中过表达(Stoica,G.等,J.Biol.Chem.,2001,276,16772-16779)。在小鼠研究中,ALK的剥夺降低成胶质细胞瘤的肿瘤生长并延长动物存活期(Powers,C.等,J.Biol.Chem.,2002,277,14153-14158;Mentlein,R.等,J.Neurochem.,2002,83,747-753)。
预计ALK抑制剂当与目前用于ALCL、IMT、增殖性障碍、成胶质细胞瘤和可能的其他实体肿瘤的化疗组合时将允许长期治愈,或者作为单一治疗剂可用于维持作用,以在那些患者中预防癌症复发。已报道了各种ALK抑制剂,例如吲唑并异喹啉类(WO 2005/009389)、噻唑酰胺类和噁唑酰胺类(WO 2005/097765)、吡咯并嘧啶类(WO 2005080393)和嘧啶二胺类(WO 2005/016894)。
WO 2008/051547公开了2,4-二氨基嘧啶的稠合双环衍生物作为ALK和c-Met抑制剂。在’547申请中公开的前导药物候选物是CEP-28122,一种在小鼠异种移植物模型中具有针对SUP-M2和Karpas-299ALK依赖性肿瘤的口服疗效的强效ALK抑制剂。CEP-28122进展到IND授权研究,直到它的开发由于在CEP-28122治疗的猴中出人意料地发生严重肺毒性而被终止。
粘着斑激酶(FAK)是位于粘着斑、即细胞与ECM(细胞外基质)的接触位点处的进化上保守的非受体酪氨酸激酶,起到来自于整合蛋白受体和多种受体酪氨酸激酶包括EGF-R、HER2、IGF-R1、PDGF-R和VEGF-R2以及TIE-2的信号的关键转导物的作用(Parsons,JT;Slack-Davis,J;Tilghman,R;Roberts,WG.“粘着斑激酶:靶向粘附信号传导途径以用于治疗性干预”(Focal adhesion kinase:targeting adhesion signaling pathways for therapeutic intervention),Clin.Cancer Res.,2008,14,627-632;Kyu-Ho Han,E;McGonigal,T.,“粘着斑激酶在人类癌症中的作用——用于药物发现的潜在靶点”(Role of focal adhesion kinase in human cancer-a potential target for drug discovery),Anti-cancer Agents Med.Chem.,2007,7,681-684)。整合蛋白激活的FAK与Src形成二元复合物,其可以磷酸化其他底物并触发多个信号传导途径。鉴于FAK结合和磷酸化在利用多种SH2-和SH3-结构域效应蛋白介导信号转导中的中心作用(Mitra,SK;Hanson,DA;Schlaeper,DD.,“粘着斑激酶:细胞运动的命令和控制”(Focal adhesion kinase:in command and control of cell motility),Nature Rev.Mol.Cell Biol.,2005,6,56-68),在正常和恶性细胞中,活化的FAK在介导细胞粘附、迁移、形态发生、增殖和存活中发挥中心作用(Mitra等,2005;McLean,GW;Carragher,NO;Avizzienyte,E等,“粘着斑激酶在癌症中的作用——新的治疗机会”(The role of focal adhesion kinase in cancer–a new therapeutic opportunity),Nature Reviews Cancer,2005,5,505-515;以及Kyu-Ho Han和McGonigal,2007)。在肿瘤中,FAK的活化介导不依赖于锚定的细胞存活,这是癌细胞的标志之一。此外,FAK过表达和活化似乎与这些恶性肿瘤中增强的侵入性和转移性表型和肿瘤血管发生相关(Owens,LV;Xu,L;Craven,RJ等,“粘着斑激酶(p125FAK)在侵入性人类肿瘤中的过表达”(Over expression of the focal adhesion kinase(p125FAK)in invasive human tumors),Cancer Res.,1995,55,2752-2755;Tremblay,L;Hauck,W.,“在人类转移性前列腺癌中粘着斑激酶(pp125FAK)的表达、活化以及与桩蛋白和p50CSK的关联”(Focal adhesion kinase(pp125FAK)expression,activation and association with paxillin and p50CSK in human metastatic prostate carcinoma),Int.J.Cancer,1996,68,164-171;Kornberg,IJ.,“口腔癌中的粘着斑激酶”(Focal adhesion kinase in oral cancers),Head and Neck,1998,20:634-639;Mc Clean等,2005;Kyu-Ho Han和McGonigal,2007),并且与不良预后和更短的无转移存活相关。
使用siRNA(Halder,J;Kamat,AA;Landen,CN等,“使用中性脂质体中的体内短干扰RNA递送进行的粘着斑激酶用于卵巢癌的治疗”(Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy),Clin.Cancer Res.,2006,12,4916-4924)、显性失活的FAK和小分子FAK抑制剂(Halder,J;Lin,YG;Merritt,WM等,“新的粘着斑激酶抑制剂TAE226在卵巢癌中的治疗效能”(Therapeutic efficacy of a novel focal adhesion kinase inhibitor,TAE226in ovarian carcinoma),Cancer Res.,2007,67,10976-10983;Roberts,WG;Ung,E;Whalen,P等,“选择性粘着斑激酶抑制剂PF-562,271的抗肿瘤活性和药理学”(Anti-tumor activity and pharmacology of a selective focal adhesion kinase inhibitor,PF-562,271),Cancer Res.,2008,68,1935-1944;Bagi CM;Roberts GW和Andersen CJ,“粘着斑激酶/Pyk2双重抑制剂对骨肿瘤具有正面效果——骨转移肿瘤的影响”(Dual focal adhesion kinse/Pyk2inhibitor has positive effects on bone tumors-implications for bone metastases),Cancer,2008,112,2313-2321)在各种实体肿瘤中进行的多项概念验证研究,为FAK抑制作为抗肿瘤/抗血管发生策略、尤其是用于雄激素不依赖性前列腺癌、乳腺癌和HNSCC的治疗性用途,提供了临床前支持。在裸大鼠中的人类乳腺癌的临床前模型(MDA-MB-231)中,小分子FAK抑制剂(PF-562,271)的给药抑制原发肿瘤生长和胫骨内肿瘤扩散,并恢复肿瘤诱导的骨损失(Bagi等,2008)。Roberts等,(2008)显示了PF-562,271抑制骨转移肿瘤,防止骨再吸收,并在出现和没有出现骨转移的乳腺癌和雄激素不依赖性前列腺癌患者中提高骨发生,支持了FAK抑制在这些特定恶性肿瘤中的其他益处。
概括来说,存在明显的遗传学和生物学证据将异常ALK活化和FAK的组成性活化与人类中某些类型癌症的发生和发展相联系。相当多的证据表明,ALK和FAK阳性肿瘤细胞需要这些癌基因才能增殖和存活,并且在FAK的情况下,才能侵入并转移到远处位点,而ALK和FAK信号传导两者的抑制导致肿瘤细胞生长停止或凋亡,造成目标的细胞减少效果。FAK的抑制还引起肿瘤移动性、侵入性和转移扩散的减弱,尤其是在以骨转移散播和溶骨病变为特征的特定癌症中。FAK活化保护肿瘤细胞免于化疗诱导的凋亡,有助于肿瘤耐药性;FAK活性的调节(通过siRNA或在药理上)增强化疗药剂(例如阿霉素、多西紫杉醇和吉西他滨)在体内的效能,表明了合理的组合疗法在特定癌症中的用途。ALK和FAK在健康成年人中的大多数正常组织中表达极少,并且在特定癌症中,在肿瘤发生期间和/或恶性发展的早期阶段中被激活和/或失调。因此,使用ALK和FAK双重抑制剂的治疗针对正常细胞的命中效果应该最小化,产生有利的治疗指数。
对用于癌症治疗的其他安全有效的ALK和/或FAK抑制剂,存在着需求。
技术实现要素:
本发明提供了式(I)的化合物
或其盐形式。
所述式(I)的化合物具有ALK和FAK抑制活性,并且可用于治疗ALK或FAK介导的障碍或病症。
本发明还提供了包含本发明的至少一种化合物和至少一种可药用赋形剂的药物组合物。
附图说明
图1示出了用于制备CEP-37440的过程的示意图。
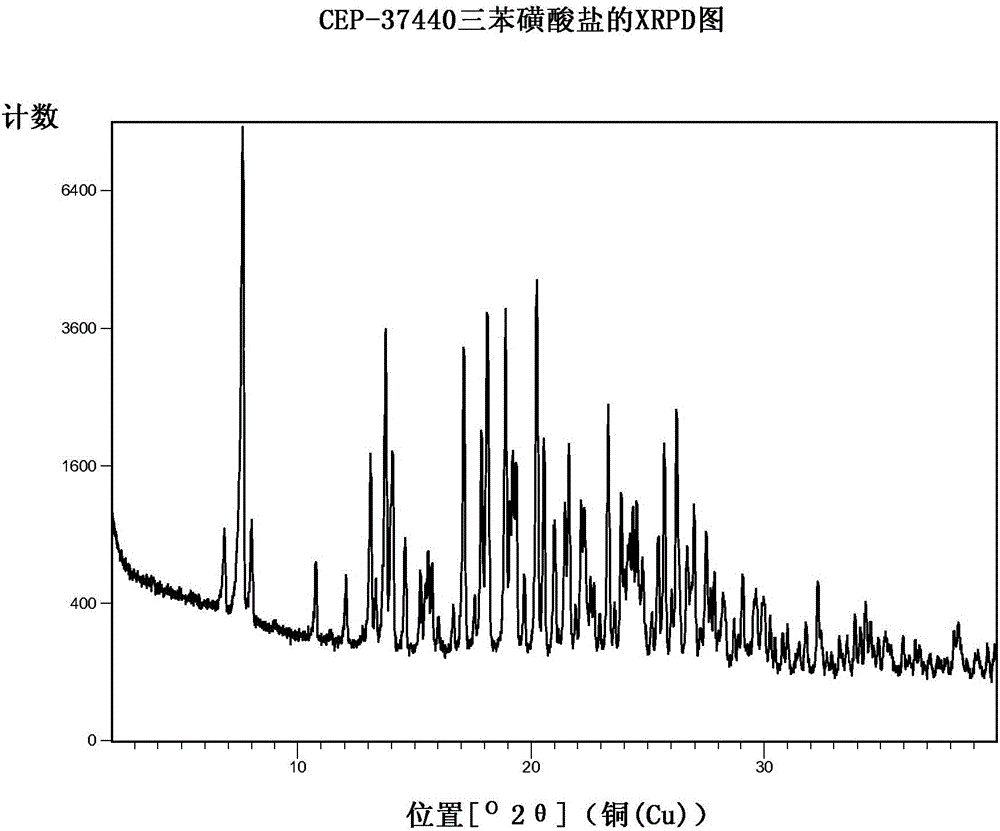
图2示出了结晶的CEP-37440三苯磺酸盐的XRPD图。
图3示出了结晶的CEP-37440二水合三盐酸盐的XRPD图。
图4示出了口服CEP-37440在小鼠中的Sup-M2ALCL肿瘤异种移植物中的抗肿瘤效能。
图5示出了用CEP-37440口服给药的带有Sup-M2ALCL肿瘤异种移植物的小鼠的体重。
图6示出了在口服给药后,在带有Sup-M2ALCL肿瘤异种移植物的小鼠中CEP-37440的血浆和肿瘤水平。
图7示出了口服CEP-37440在小鼠中的Karpas-299肿瘤异种移植物中的抗肿瘤效能。
图8示出了用CEP-37440口服给药的带有Karpas-299肿瘤异种移植物的小鼠的体重。
图9示出了在口服给药后,在带有Karpas-299肿瘤异种移植物的小鼠中CEP-37440的血浆和肿瘤水平。
图10示出了口服CEP-37440在小鼠中的NCI-H2228NSCL肿瘤异种移植物中的抗肿瘤效能。
图11示出了口服CEP-37440在小鼠中的NCI-H3122NSCL肿瘤异种移植物中的抗肿瘤效能。
图12示出了在口服给药后,CEP-37440在带有NCI-H2228NSCL肿瘤异种移植物的小鼠中的血浆和肿瘤水平。
图13示出了在口服给药后,CEP-37440在带有NCI-H3122NSCL肿瘤异种移植物的小鼠中的血浆和肿瘤水平。
图14示出了用CEP-37440口服给药的带有NCI-H2228NSCL肿瘤异种移植物的小鼠的体重。
图15示出了用CEP-37440口服给药的带有NCI-H3122NSCL肿瘤异种移植物的小鼠的体重。
图16示出了口服CEP-37440在小鼠中的NCI-H2228NSCL肿瘤异种移植物中的抗肿瘤效能和长期肿瘤减退。
图17示出了用CEP-37440口服给药的带有NCI-H2228NSCL肿瘤异种移植物的小鼠的体重。
图18示出了口服CEP-37440和PF-562271在带有PC-3前列腺肿瘤异种移植物并具有组成性FAK活化的裸小鼠中的抗肿瘤效能。
图19示出了口服CEP-37440和PF-562271在带有HCC-827人类NSCL癌异种移植物(EML4-ALK阴性)的裸小鼠中的抗肿瘤效能。
图20示出了用CEP-37440或PF-562271口服给药的带有HCC-827人类NSCL癌异种移植物(EML4-ALK阴性)的裸小鼠的体重。
图21示出了FAK的肿瘤药效学抑制和口服CEP-37440和PF-562271在带有Detroit 562人类HNSCC癌异种移植物(EML4-ALK阴性)的SCID小鼠中的抗肿瘤效能。
发明详述
I.定义
当在本文中使用时,除非另有规定,否则下列术语具有赋予它们的含义。
“药物组合物”是指具有适合给药于人类的安全性/效能情况的组合物。
“可药用赋形剂”是指生理上可耐受的材料,其在给药于人类时通常不产生过敏或其他不良反应例如胃部不适、晕眩等。
“可药用盐”是指具有适合给药于人类的安全性/效能情况的盐。
“受试者”是指哺乳动物纲的成员。哺乳动物的实例包括但不限于人类、灵长动物、黑猩猩、啮齿动物、小鼠、大鼠、兔、马、家畜、狗、猫、绵羊和奶牛。
“治疗有效量”是指足以在特定受试者或受试者群体中改善或抑制与待治疗的障碍或病症相关的症状的恶化的化合物的量。应该认识到,适合的剂型、剂量和给药途径的确定在制药和医药领域的普通技术人员的水平之内。
“治疗”是指与待治疗的障碍相关或由其引起的至少一种症状或特征的快速或预防性减轻或缓解。例如,治疗可以包括障碍症状的减轻或障碍的完全根除。
II.化合物
本发明提供了式(I)的化合物
或其盐形式。式(I)的化合物具有化学名2-[[5-氯-2-[[(6S)-6-[4-(2-羟乙基)哌嗪-1-基]-1-甲氧基-6,7,8,9-四氢-5H-苯并[7]轮烯-2-基]氨基]嘧啶-4-基]氨基]-N-甲基-苯甲酰胺,并且也被称为CEP-37440。我们已发现,式(I)的化合物与CEP-28122和其他相关化合物相比具有令人吃惊和出人意料的性质。
式(I)的化合物的盐形式优选为可药用的。式(I)的化合物的可药用酸盐形式包括但不限于源自于无机酸例如盐酸、硝酸、磷酸、硫酸、氢溴酸、氢碘酸、亚磷酸的盐及它们的混合物,以及源自于有机酸例如脂族单羧酸和二羧酸、苯基取代的烷酸、羟基烷酸、烷二酸、芳香族酸和脂族和芳香族磺酸的盐。因此,这样的酸盐包括但不限于硫酸盐、焦硫酸盐、硫酸氢盐、亚硫酸盐、亚硫酸氢盐、硝酸盐、磷酸盐、磷酸单氢盐、磷酸二氢盐、偏磷酸盐、焦磷酸盐、氯化物、溴化物、碘化物、乙酸盐、丙酸盐、辛酸盐、异丁酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、延胡索酸盐、马来酸盐、扁桃酸盐、苯甲酸盐、氯代苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、邻苯二甲酸盐、苯磺酸盐、甲苯磺酸盐、苯乙酸盐、柠檬酸盐、乳酸盐、马来酸盐、酒石酸盐、甲磺酸盐及它们的混合物。在某些实施方式中,酸盐选自苯磺酸盐和氯化物。在某些实施方式中,酸盐是氯化物。在某些实施方式中,酸盐是三苯磺酸盐。在某些实施方式中,三苯磺酸盐的特征在于XRPD图具有选自7.62、13.11、13.76和14.05±0.2°2θ的一个或多个峰。在某些实施方式中,三苯磺酸盐的特征在于XRPD图具有选自6.85、7.62、8.01、13.11、13.76、14.05和14.60±0.2°2θ的一个或多个峰。在某些实施方式中,三苯磺酸盐的特征在于XRPD图具有选自7.62、13.11、13.76、14.05、17.10、17.86和18.10±0.2°2θ的一个或多个峰。在某些实施方式中,酸盐是三盐酸盐。在某些实施方式中,酸盐是二水合三盐酸盐。在某些实施方式中,二水合三盐酸盐的特征在于XRPD图具有选自5.42、8.86、14.06、17.52和18.51±0.2°2θ的一个或多个峰。在某些实施方式中,二水合三盐酸盐的特征在于XRPD图具有选自5.42、5.91、8.86、10.80、11.79、14.06、14.72、17.02、17.52和18.51±0.2°2θ的一个或多个峰。在某些实施方式中,酸盐是单水合三盐酸盐。
酸加成盐可以通过以常规方式将式(I)的化合物与足够量的所需酸接触以生产盐来制备。式(I)的化合物的游离碱形式可以通过以常规方式将盐形式与碱接触并分离游离碱来再生。
本发明包括以任何物理形式、包括无定形或以任何多晶形式的结晶固体,处于任何纯度状态的式(I)的化合物及其盐形式。结晶多晶形式包括非溶剂化形式以及溶剂化形式,例如水合形式。制备有机化合物例如式(I)的化合物的结晶和无定形形式的方法,对于本领域普通技术人员来说是公知的。
III.药物组合物
本发明还提供包含本发明的化合物(例如式(I)的化合物或其可药用盐)和可药用赋形剂在一起的药物组合物。为了从本发明的化合物制备药物组合物,可药用赋形剂可以是固体或液体。赋形剂可以是可能作为例如载体、稀释剂、调味剂、粘合剂、防腐剂、片剂崩解剂或包封材料起作用的一种或多种物质。药物组合物可以含有两种或更多种本发明的化合物(例如式(I)的化合物的两种或更多种不同盐形式)。优选地,药物组合物含有治疗有效量的式(I)的化合物或其可药用盐形式。在一种实施方式中,组合物含有有效治疗ALK或FAK介导的障碍或病症的量的式(I)的化合物或其可药用盐形式。优选地,当定量或定性测量时,本发明的药物组合物引起与ALK或FAK介导的障碍相关的症状或疾病迹象的减轻。除了式(I)的化合物或其可药用盐形式和可药用赋形剂之外,组合物还可以包含另一种治疗性化合物,例如在癌症治疗中有用的化合物。
本发明的化合物可以配制成任何形式的药物组合物,例如糖浆、酏剂、悬液、粉剂、颗粒剂、片剂、胶囊、含片、锭剂、水性溶液、霜剂、软膏、洗剂、凝胶、乳液等。固体形式的制剂包括粉剂、片剂、丸剂、胶囊、扁囊剂、栓剂和可分散颗粒剂。优选地,药物组合物是片剂或胶囊。在一种实施方式中,药物组合物是片剂。在另一种实施方式中,药物组合物是胶囊。
在粉剂中,赋形剂可以是与细分活性组分混合的细分固体。在片剂中,可以将活性组分与具有必需的粘合性质的赋形剂以适合比例混合,并压制成所需形状和尺寸。适合的赋形剂包括碳酸镁、硬脂酸镁、滑石粉、糖、乳糖、果胶、糊精、淀粉、明胶、黄耆树胶、甲基纤维素、羧甲基纤维素钠、低熔点蜡、可可脂等。
药物组合物优选地含有1%至95%(w/w)的活性化合物。更优选地,药物组合物含有5%至70%(w/w)的活性化合物。
为了制备栓剂,可以将低熔点蜡例如脂肪酸甘油酯的混合物或可可脂熔化,并例如通过搅拌将活性组分均匀地分散在其中。然后可以将熔融的均匀混合物倾倒在常规尺寸的模具中,使其冷却并因此固化。
液体形式的制剂包括溶液、悬液和乳液。适合于肠胃外给药,例如通过静脉内、肌肉内、真皮内和皮下途径给药的制剂,包括水性和非水性的等渗无菌注射溶液,其可以含有抗氧化剂、缓冲剂、抑菌剂和使所述制剂与目标受体的血液等渗的溶质,以及水性和非水性无菌悬液,其可以包含悬浮剂、增溶剂、增稠剂、稳定剂和防腐剂。在本发明的实践中,组合物可以例如通过静脉内输注、口服、表面、腹膜内、膀胱内或鞘内方式给药。化合物的制剂可以存在于单位剂量或多剂量密封容器例如安瓿和小瓶中。注射溶液和悬液可以从上面描述类型的无菌粉剂、颗粒剂和片剂来制备。
单独或与其他适合组分组合的本发明的化合物,可以被制造成气溶胶制剂(即它们可以被“雾化”),以通过吸入来给药。气溶胶制剂可以放置在加压的可接受推进剂例如二氯二氟甲烷、丙烷、氮气等中。
可药用赋形剂部分由待给药的具体组合物以及由用于给药所述组合物的具体方法所决定。因此,本发明的药物组合物存在广泛的各种适合制剂(参见例如《Remington药物学科学与实践》(Remington:The Science and Practice of Pharmac),第20版,Gennaro等主编,Lippincott Williams and Wilkins,2000)。
根据具体应用,药物组合物中活性组分的量可以被改变或调整,例如1mg至1,000mg、5mg至500mg、10mg至300mg或25mg至250mg。
给药于受试者的剂量优选地足以在受试者中随时间引起有益的治疗响应。有益的剂量可以随受试者而变,取决于例如受试者的状况、体重、表面积和副作用易感性。给药可以通过单次剂量或分次剂量来实现。
IV.治疗方法
另一方面,本发明提供了在受试者中治疗ALK或FAK介导的障碍或病症的方法,所述方法包括:向受试者给药式(I)的化合物或其可药用盐形式。另一方面,本发明提供了用于在受试者中治疗ALK或FAK介导的障碍或病症的式(I)的化合物或其可药用盐形式。另一方面,本发明提供了式(I)的化合物或其可药用盐形式,其用于制备治疗受试者中ALK或FAK介导的障碍或病症的药物。优选地,将式(I)的化合物或其可药用盐形式作为包含可药用赋形剂的药物组合物给药于受试者。优选地,式(I)的化合物或其可药用盐形式以治疗有效量给药于受试者。在一种实施方式中,ALK或FAK介导的病症或障碍是癌症。在另一种实施方式中,ALK或FAK介导的病症或障碍选自间变性大细胞淋巴瘤(ALCL)、非小细胞肺癌(NSCLC)、成神经细胞瘤、成胶质细胞瘤、前列腺癌、鳞状细胞癌(SCC)和乳腺癌。在某些实施方式中,ALK或FAK介导的病症或障碍选自ALK阳性ALCL、EML4-ALK阳性NSCLC、成神经细胞瘤、成胶质细胞瘤、雄激素不依赖性前列腺癌、乳腺癌和头颈部鳞状细胞癌(HNSCC)。在某些实施方式中,ALK或FAK介导的病症或障碍选自ALK阳性ALCL、EML4-ALK阳性NSCLC、成神经细胞瘤、雄激素不依赖性前列腺癌、乳腺癌和HNSCC。在某些实施方式中,ALK或FAK介导的病症或障碍选自ALK阳性ALCL、EML4-ALK阳性NSCLC、成神经细胞瘤和成胶质细胞瘤。在某些实施方式中,ALK或FAK介导的病症或障碍选自ALK阳性ALCL、EML4-ALK阳性NSCLC和成神经细胞瘤。在某些实施方式中,ALK或FAK介导的病症或障碍选自ALK阳性ALCL和EML4-ALK阳性NSCLC。在某些实施方式中,ALK或FAK介导的病症或障碍选自雄激素不依赖性前列腺癌、乳腺癌和HNSCC。在某些实施方式中,ALK或FAK介导的病症或障碍是ALK介导的病症或障碍。在某些实施方式中,ALK或FAK介导的病症或障碍是FAK介导的病症或障碍。在某些实施方式中,ALK或FAK介导的病症或障碍是成肌纤维细胞肿瘤。在某些实施方式中,ALK或FAK介导的病症或障碍是具有TPM3-ALK或TPM4-ALK癌基因的成肌纤维细胞肿瘤。在某些实施方式中,ALK或FAK介导的病症或障碍是具有TPM3-ALK癌基因的成肌纤维细胞肿瘤。在某些实施方式中,ALK或FAK介导的病症或障碍是具有TPM4-ALK癌基因的成肌纤维细胞肿瘤。
取决于障碍或病症的本质,ALK或FAK介导的障碍或病症可以使用本发明的化合物预防性、短期或长期治疗。优选地,在每种这些方法中,受试者是人类。
在另一种实施方式中,本发明提供了在受试者中治疗增殖性障碍的方法,所述方法包括向受试者给药式(I)的化合物或其可药用盐形式。另一方面,本发明提供了式(I)的化合物或其可药用盐形式,其用于在受试者中治疗增殖性障碍。另一方面,本发明提供了式(I)的化合物或其可药用盐形式,其用于制备在受试者中治疗增殖性障碍的药物。优选地,式(I)的化合物或其可药用盐形式在包含可药用赋形剂的药物组合物中给药于受试者。优选地,式(I)的化合物或其可药用盐形式以可药用量给药于受试者。在某些实施方式中,增殖性障碍是ALK或FAK介导的。在某些实施方式中,增殖性障碍是癌症。在某些实施方式中,增殖性障碍选自间变性大细胞淋巴瘤(ALCL)、非小细胞肺癌(NSCLC)、成神经细胞瘤、成胶质细胞瘤、前列腺癌、鳞状细胞癌(SCC)和乳腺癌。在某些实施方式中,增殖性障碍选自ALK阳性ALCL、EML4-ALK阳性NSCLC、成神经细胞瘤、成胶质细胞瘤、雄激素不依赖性前列腺癌、乳腺癌和头颈部鳞状细胞癌(HNSCC)。在某些实施方式中,增殖性障碍选自ALK阳性ALCL、EML4-ALK阳性NSCLC、成神经细胞瘤、雄激素不依赖性前列腺癌、乳腺癌和HNSCC。在某些实施方式中,增殖性障碍选自ALK阳性ALCL、EML4-ALK阳性NSCLC、成神经细胞瘤和成胶质细胞瘤。在某些实施方式中,增殖性障碍选自ALK阳性ALCLE、ML4-ALK阳性NSCLC和成神经细胞瘤。在某些实施方式中,增殖性障碍选自ALK阳性ALCL和EML4-ALK阳性NSCLC。在某些实施方式中,增殖性障碍选自雄激素不依赖性前列腺癌、乳腺癌和HNSCC。
取决于障碍或病症的本质,增殖性障碍可以使用本发明的化合物预防性、短期或长期治疗。优选地,在每种这些方法中,受试者是人类。
在治疗性应用中,本发明的化合物可以以种类繁多的口服和肠胃外剂型得以制备并给药。因此,本发明的化合物可以通过注射、即静脉内、肌肉内、真皮内、皮下、十二指肠内或腹膜内注射给药。在某些实施方式中,本发明的化合物以静脉内或皮下给药。此外,本文描述的化合物可以通过吸入例如鼻内给药。此外,本发明的化合物可以透皮给药。在另一种实施方式中,本发明的化合物经口递送。化合物还可以经直肠、颊或通过吹入法递送。
为特定情况确定适合的剂量,在从业人员的技术范围之内。一般来说,治疗从低于化合物的最适剂量的较小剂量开始。随后,通过小的增量逐步增加剂量,直至达到所述情况下的最优效果。方便起见,如果需要,每日总剂量可以分成多份并在一天中给药。典型剂量为约1mg至约1,000mg每天,例如约5mg至约500mg每天。在某些实施方式中,剂量为约10mg至约300mg每天,例如约25mg至约250mg每天。
具体实施方式
V.化学
CEP-28122
(1S,2S,3R,4R)-3-[5-氯-2-((S)-1-甲氧基-7-吗啉-4-基-6,7,8,9-四氢-5H-苯并环庚烯-2-基氨基)-嘧啶-4-基氨基]-双环[2.2.1]庚-5-烯-2-羧酸酰胺(CEP-28122)按照WO 2008/051547(Ahmed等)的实施例882中所述来制备。
CEP-37440
2-(5-氯-2-{(S)-6-[4-(2-羟基-乙基)-哌嗪-1-基]-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-2-基氨基}-嘧啶-4-基氨基)-N-甲基-苯甲酰胺的合成可以按照图1,遵照步骤1-8中概括的程序来进行。
步骤1:5-甲氧基-1-亚甲基-1,2,3,4-四氢-萘:向室温下的5-甲氧基-3,4-二氢-2H-萘-1-酮(25g,0.14mol)和甲基三苯基碘化膦(1.13eq)在THF(250mL)中的悬液加入叔丁醇钾(1.6eq),添加速率使得将温度维持在不高于摸起来温暖的水平。将反应搅拌1小时并浓缩。然后将反应与三倍体积的己烷共沸,以除去过量叔丁醇。加入新鲜己烷,将溶液静置过夜以进行研磨。通过过滤除去红棕色固体,并将滤液用水洗涤两次并浓缩。在ISCO上通过层析进行纯化(330g SiO2柱:逐步己烷、然后DCM洗脱),得到作为浅黄色油状物的标题化合物(24g,99%)。1H-NMR(400MHz,CDCl3)7.29(d,J=8.0Hz,1H),7.15(t,J=8.0Hz,1H),6.76(d,J=8.0Hz,1H),5.49(s,1H),4.98(s,1H),3.85(s,3H),2.77(t,J=6.4Hz,2H),2.53-2.50(m,2H),1.93-1.87(m,2H)。
步骤2:1-甲氧基-5,7,8,9-四氢-苯并环庚烯-6-酮:向新鲜制备的三水硝酸铊(III)(1.0eq)在300mL MeOH中的溶液一次性加入150mL MeOH中的5-甲氧基-1-亚甲基-1,2,3,4-四氢-萘(23.8g,0.137mol)。搅拌1分钟并加入400mL氯仿。将溶液过滤,并将有机物在二氯甲烷与水之间分配。将有机物干燥(MgSO4)并浓缩。通过层析进行纯化(ISCO,330g二氧化硅柱:逐步己烷洗脱(5min),然后7分钟梯度洗脱至100%二氯甲烷(20min)),得到作为极性最高的产物的标题化合物,其是浅黄色油状物(26g,97%)。1H-NMR(400MHz,CDCl3)7.16(t,J=7.9Hz,1H),7.84(d,J=8.3Hz,1H),6.79(d,J=7.5Hz,1H),3.84(s,3H),3.73(s,2H),3.05-3.01(m,2H),2.55(t,J=7.0Hz,2H),2.01-1.96(m,2H)。LC/MS(ESI+)m/z=191(M+H)+。
步骤3:1-甲氧基-2-硝基-5,7,8,9-四氢-苯并环庚烯-6-酮:在0℃下,向乙腈(50mL)和三氟乙酸酐(100mL)中的硝酸钾逐滴加入50mL乙腈中的1-甲氧基-5,7,8,9-四氢-苯并环庚烯-6-酮(25g,0.131mol)。在升温至室温的同时将反应搅拌2.5小时。在旋转蒸发仪上不加热将反应浓缩。加入MeOH并简短搅拌。重新浓缩并通过在二氯甲烷与碳酸氢钠饱和水溶液之间分配进行精制。分离有机层并干燥(Mg2SO4)、浓缩,通过层析ISCO进行纯化(330g二氧化硅柱:梯度洗脱-在60分钟内10至50%EA:HEX),得到两种异构体。标题化合物被较晚洗脱(10.7克,得率为34.6%)。1H-NMR(400MHz,CDCl3)7.70(d,J=8.3Hz,1H),7.06(d,J=8.3Hz,1H),3.92(s,3H),3.80(s,2H),3.13-3.09(m,2H),2.60(t,J=7.0Hz,2H),2.10-2.03(m,2H)。
步骤4:2-[4-(1-甲氧基-2-硝基-6,7,8,9-四氢-5H-苯并环庚烯-6-基)-哌嗪-1-基]-乙醇:将二氯甲烷(870ml)中的1-甲氧基-2-硝基-5,7,8,9-四氢-苯并环庚烯-6-酮(15.09g,64.15mmol)用2-哌嗪-1-基-乙醇(3eq)、然后用乙酸(10eq)处理。将混合物在50℃下搅拌2小时,冷却至0℃,加入三乙酰氧基硼氢化钠(4eq),然后升温至室温并搅拌。在几小时后,起始原料仍然存在。另外加入0.4eq三乙酰氧基硼氢化钠,然后在6小时后再次重复。搅拌过夜。倾倒在碳酸氢钠饱和水溶液和冰中,并用1N氢氧化钠制成碱性到pH 10,用二氯甲烷萃取两次,MgSO4干燥,过滤并浓缩。将该材料转移到乙醇中,并加入HCl/乙醇。将得到的沉淀研磨2小时,然后过滤。使用NaOH然后用碳酸氢钠使固体成为游离碱,并萃取到二氯甲烷中,得到标题化合物(19g,85%得率)。1H-NMR(400MHz,CDCl3)7.56(d,J=8.2Hz,1H),7.00(d,J=8.2Hz,1H),3.82(s,3H),3.63-3.06(m,2H),3.29-3.24(m,1H),3.00-2.86(m,3H),2.72-2.67(m,2H),2.60-2.51(m,8H),2.46-2.37(m,2H),2.12-2.07(m,2H),1.87-1.78(m,1H),1.37-1.29(m,1H)。LC/MS(ESI+)m/z=350(M+H)+。
步骤5:2-[4-(2-氨基-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-6-基)-哌嗪-1-基]-乙醇:将2-[4-(1-甲氧基-2-硝基-6,7,8,9-四氢-5H-苯并环庚烯-6-基)-哌嗪-1-基]-乙醇(19.0g,54.4mmol)分成两批,并溶解在乙醇(总共232mL)中。将10%Pd/C(1.74g,1.64mmol)分为两半,并将反应在50psi下加氢3-4小时。将每个反应混合物通过硅藻土过滤以除去Pd。将滤液合并然后浓缩,分离到作为泡沫状固体的标题化合物(17.25g,99%得率)。1H-NMR(400MHz,CDCl3)6.76(d,J=7.9Hz,1H),6.53(d,J=7.9Hz,1H),3.72(宽单峰,3H),3.71(s,3H),3.64(t,J=5.4Hz,2H),3.26-3.20(m,1H),2.84-2.72(m,5H),2.62-2.56(m,8H),2.42-2.35(m,2H),2.40-2.37(m,1H),1.81-1.74(m,1H),1.70(宽单峰,1H),1.41-1.33(m,1H)。LC/MS(ESI+)m/z=320(M+H)+。
步骤6:2-[4-((S)-2-氨基-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-6-基)-哌嗪-1-基]-乙醇:将34克消旋的2-[4-(2-氨基-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-6-基)-哌嗪-1-基]-乙醇使用SFC(超临界流体CO2)层析进行分离,使用Chiralcel OJ-H(3x15cm)808041柱和15%甲醇(0.2%DEA)/CO2,以80mL/min流速在100bar下洗脱,监测220nm波长,进样体积:0.5mL,20mg/mL乙醇。分离到16.9克(R)-对映异构体和17克标题化合物,化学纯度>99%,对映异构过量值(ee)>99%(使用Chiralcel OJ-H分析柱测量)。NMR和质量与消旋材料等同。通过小分子X-射线,使用双对溴苯甲基衍生物:4-溴-苯甲酸2-{4-[(R)-2-(4-溴-苯甲基氨基)-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-6-基]-哌嗪-1-基}-乙基酯的不规则分散系,第一洗脱异构体的绝对构型被明确指定为(R)-构型。因此,第二洗脱对映异构体被确定为(S)-构型。
步骤7:2-(2,5-二氯-嘧啶-4-基氨基)-N-甲基-苯甲酰胺:向DMF(0.5L)中的2-氨基-N-甲基-苯甲酰胺(24.4g,0.16mol)加入2,4,5-三氯-嘧啶(39g,1.3eq)和碳酸钾(1.3eq)。在氩气下在75℃搅拌5小时,然后在室温下搅拌过夜。倾倒在1L水中,通过过滤分离沉淀物并用1:1的乙腈:水洗涤,然后在空气流中和真空下干燥,得到作为黄色固体的标题化合物(38g,78%得率)。11.70(s,1H),8.74(d,J=8.2Hz,1H),8.24(s,1H),7.59(d,J=8.4Hz,1H),7.53(d,J=8.8Hz,1H),7.16(t,J=8.4Hz,1H),6.28(s,1H),3.06(d,J=4.7Hz,3H)。
步骤8:2-(5-氯-2-{(S)-6-[4-(2-羟基-乙基)-哌嗪-1-基]-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-2-基氨基}-嘧啶-4-基氨基)-N-甲基-苯甲酰胺:在密封容器中,将2-[4-((S)-2-氨基-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-6-基)-哌嗪-1-基]-乙醇(2.69g,8.41mmol)和2-(2,5-二氯-嘧啶-4-基氨基)-N-甲基-苯甲酰胺(2.00g,6.73mmol)合并在1-甲氧基-2-丙醇(120mL,1200mmol)中,然后加入甲磺酸(2.44mL,37.7mmol)。然后将反应在90℃加热18小时。
将反应混合物加入到分液漏斗中,用饱和碳酸氢钠溶液稀释,直至形成云雾状沉淀。将它用二氯甲烷萃取3次。然后将有机层用盐水洗涤,在MgSO4上干燥,过滤并浓缩。将残留物泵干,然后在ISCO快速柱上层析。将它注入到正相柱上的二氯甲烷中,在0-10%(二氯甲烷:MeOH中的10%NH4OH)的梯度上洗脱。所需产物在9-10%左右洗脱,保持10%的梯度直至产物被完全洗脱。将混合的级分浓缩,并在Gilson反相HPLC上层析,使用0-40%CH3CN进行梯度洗脱。根据需要,使用正相二氧化硅和反相HPLC重复层析,以执行进一步纯化。在中和并浓缩所有材料后,通过将泡沫转移到EtOAc中并浓缩至干几次来获得得到的固体,以得到标题化合物(1.1g,28%)。11.02(s,1H),8.69(d,J=8.9Hz,1H),8.13(s,1H),8.08(d,J=8.4Hz,1H),7.59-7.50(m,2H),7.41(s,1H),7.13(t,J=7.4Hz,1H),6.91(d,J=8.1Hz,1H),6.21(s,1H),3.74(m,3H),3.66-3.63(m,2H),3.29-3.23(m,1H),3.06(d,J=4.3Hz,3H),2.92-2.72(m,5H),2.66-2.55(m,8H),2.48-2.39(m,2H),2.16-2.10(m,2H),1.87-1.77(m,1H),1.42-1.32(m,1H)。LC/MS(ESI+)m/z=580(M+H)+。
CEP-37440无定形HCl盐
2-(5-氯-2-{6-[4-(2-羟基-乙基)-哌嗪-1-基]-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-2-基氨基}-嘧啶-4-基氨基)-N-甲基-苯甲酰胺盐酸盐:将2-(5-氯-2-{6-[4-(2-羟基-乙基)-哌嗪-1-基]-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-2-基氨基}-嘧啶-4-基氨基)-N-甲基-苯甲酰胺(4.90g,8.45mmol)和乙醇(13.5mL,33.8mmol)中的2.5M HCl加热,直至它们溶解在乙醇(164mL)中。将反应从乙醇浓缩两次,然后在少量乙醇中加温直至完全溶解。使该溶液在搅拌(<100rpm)下缓慢冷却。在溶液冷却之前,固体沉淀物快速形成。将该混合物搅拌直至达到室温,然后过滤。将固体用乙醇洗涤,然后用乙醚洗涤,随后在高真空下直接泵干,得到2-(5-氯-2-{6-[4-(2-羟基-乙基)-哌嗪-1-基]-1-甲氧基-6,7,8,9-四氢-5H-苯并环庚烯-2-基氨基}-嘧啶-4-基氨基)-N-甲基-苯甲酰胺盐酸盐(5.3克,定量得率)。1H-NMR(MeOD,400MHz)δ8.55(s,1H),8.17(s,1H),7.80(d,J=7.7Hz,1H),7.55(t,J=6.8Hz,1H),7.46(宽单峰,1H),7.36(t,J=7.2Hz,1H),7.23(d,J=8.5Hz,1H),4.00-3.95(m,4H),3.83-3.72(m,5H),3.73(s,3H),3.65-3.59(m,2H),3.47-3.38(m,5H),2.95(s,3H),2.72-2.65(m,1H),2.44-2.38(m,1H),2.29-2.28(m,1H),2.19-2.12(m,1H),1.59-1.49(m,1H)。LC/MS(ESI+)m/z=580(M+H)+。
CEP-37440盐筛选
进行了下列盐筛选实验:(a)在甲醇中,使用27种不同的酸(乙酸,抗坏血酸,苯磺酸,柠檬酸,D-葡萄糖醛酸,DL-谷氨酸,DL-乳酸,乙烷-1,2-二磺酸,乙磺酸,延胡索酸,乙醇酸,马尿酸,氢溴酸(48%水溶液),盐酸(2.5M,EtOH),L-天冬氨酸,L-酒石酸,L-焦谷氨酸,乳糖酸,马来酸,苹果酸,丙二酸,萘-2-磺酸,正磷酸,单水对甲苯磺酸,丙酸,琥珀酸和硫酸),(b)在二氯甲烷和四氢呋喃中,使用9种不同的酸(苯磺酸,乙烷-1,2-二磺酸,乙磺酸,氢溴酸(48%水溶液),萘-2-磺酸,正磷酸(85%),硫酸,对甲苯磺酸和盐酸(乙醇)),以及(c)在氯仿、丙酮、乙酸乙酯和1-丙醇中,使用苯磺酸、氢溴酸(48%水溶液)、正磷酸、硫酸、对甲苯磺酸、和盐酸(乙醇)。尽管许多实验提供了盐形式,但仅有几个实验产生了结晶盐,并且仅有两种盐形式是结晶、稳定和可重现的:三苯磺酸盐形式和二水合三盐酸盐形式。从制药前景来看,二水合三盐酸盐比三苯磺酸盐有利,因为HCl是公认安全的(GRAS)1类酸加成盐,而苯磺酸是非GRAS的2类酸加成盐(参见Stahl,H.P.&Wermuth,C.G.主编,2002《制药用盐手册:性质、选择和用途》(Handbook of Pharmaceutical salts;Properties,Selection,and Use),Vwerlag HelveticaChimica Acta and Wiley-VCH)。除了毒性更低之外,HCl具有比苯磺酸更低的分子量,这提供了更高的API/酸比率和更低的有效剂量。
在装备有X'celerator检测器的PANalytical X Pert Pro衍射仪上,使用45kV和40mA下的CuKα辐射记录粉末X-射线衍射图。Kα1辐射使用高度定向晶体(Ge111)入射光束单色器来获得。将10mm光束罩和固定(1/4°)发散和抗散射(1/8°)狭缝插入到入射光束一侧上。将固定的5mm接收狭缝和0.04弧度Soller块插入到衍射的光束一侧上。从约2至40°2θ,使用产生约0.5°/min的扫描速率的0.0080°的步级尺寸和96.06sec计数时间,来收集X-射线粉末图扫描。将样品铺展在用于测量的硅零背景(ZBG)板上。使用PANalytical PW3064旋转器(15转/min.)来旋转样品。在数据收集之前Si参比标准品的测量,产生良好地位于28.44<2θ<28.50的公差之内并明显高于150cps的最小峰高的2θ和强度值。
CEP-37440三苯磺酸盐
通过在室温下搅拌5分钟,将CEP-37440游离碱(500mg,0.862mmol)溶解在6mL四氢呋喃中。向20mL玻璃闪烁管(16x60mm)中的苯磺酸(545.6mg,3.45mmol)一次加入1mL这种溶液。在添加期间使用搅拌棒混合样品(在室温下)。当加入所有CEP-37440溶液时,注意到油状物加上固体。将样品在HEL PolyblockTM装置上在5-7℃下搅拌19小时。通过吸滤分离固体。将固体在低真空(house vacuum)下在50℃干燥5小时,得到835mg(80%回收率)浅黄色固体。通过将616mg悬浮在1.5mL水中并将得到的悬液在室温下搅拌30分钟,来改善HPLC纯度、含量测定和化合物颜色。通过吸滤分离固体,并将过滤垫上的固体用1mL水洗涤。将白色固体在低真空(house vacuum)下在50℃干燥68小时,得到406mg(67%回收率)。所述三苯磺酸盐具有约20-33mg/mL的水溶性。
结晶三苯磺酸盐的X-射线衍射数据特征示出在表1和图2中。
表1CEP-37440三苯磺酸盐的XRPD图的较高相对强度以及2θ位置和d-间距
最高峰值(强度100%)用粗体字母设定。
CEP-37440二水合三盐酸盐
在含有200mg CEP-37440游离碱的20mL闪烁管中,在室温下加入正丁醇(5mL)。在搅拌5分钟后,游离碱溶解。然后加入HCl(2.5M,在EtOH中,0.435mL,3.15eq),导致白色固体(无定形HCl盐)的立即沉淀。将得到的悬液在20分钟内加热至85℃。(注意:在约60℃下,没有固体残留在溶液之外)。在80至85℃之间搅拌溶液,在30分钟后引起自成核,导致白色固体的进一步沉淀。在将反应搅拌总共2小时后,将得到的悬液在1小时内冷却至5℃。将0-5℃的悬液继续搅拌1小时,然后过滤,用极少冷正丁醇洗涤。然后将固体在55℃下干燥过夜。在相对湿度30-70%的空气中静置后,获得208mgCEP-37440-3HCl-2H2O。
结晶的二水合三盐酸盐的X-射线衍射图特征示出在表2和图3中。所述盐在30-80%相对湿度下稳定,但是在低于30%相对湿度下可逆地转变成单水合三盐酸盐形式,并且在高于80%相对湿度下不可逆地转变成高度水合的无定形形式。
表2CEP-37440二水合三盐酸盐的XRPD图的较高相对强度以及2θ位置和d-间距
最高峰值(强度100%)用粗体字母设定。
VI.生物学
CEP-28122在Sprague-Dawly大鼠中的13周口服毒性和毒物动力学研究,包括4周的恢复期
向20只大鼠/性别的三个治疗组以相当于30、75和150mg游离碱/kg/天的剂量水平给药CEP-28122(作为单甲磺酸单HCl盐形式给药)。将20只动物/性别的一个另外的组用作对照并接受介质蒸馏水。药物或介质通过经口管饲法,以10mL/kg/剂的药剂体积,每天一次共连续91天给药于所有组。在给药时间段后,将5只动物/性别/组维持4周的恢复期。此外,将一组3只动物/性别和三组9只动物/性别/组用作毒物动力学(TK)动物,并以与主研究组相同的方式和相同的剂量水平接受介质或药物。
对于所有动物,每天至少两次进行发病率、死亡率、受伤和食物与水的利用性的观察。每周在主研究动物上进行临床观察。在给药开始之前(-1周)和研究期间每周,测量并记录所有动物的体重。每周测量并记录主研究动物的食物消耗量。在试验前以及终末期和恢复期尸检之前,对主研究动物进行检眼镜检查。在第4周结束时和终末期和恢复期尸检时从主研究动物收集用于指定的临床病理学评估的血样。在终末期和恢复期尸检之前从主研究动物收集用于尿液分析评估的尿样。在第1天、第4周和第13周的指定时间点,从指定的TK动物收集用于确定药物的血浆浓度的血样。在最终的血液收集之后,将TK动物安乐死并将尸体丢弃。在终末期和恢复期结束时,进行尸检,记录器官重量,并对选定的组织进行显微镜检查。
CEP-28122在食蟹猴中的13周口服毒性和毒物动力学研究,包括6周的恢复期
通过将CEP-28122单甲磺酸单HCl盐溶解在注射用无菌水(SWFI)中以达到所需药剂浓度水平,每周制备CEP-28122溶液。
将40只实验用未接触过抗原的食蟹猴(20只雄性和20只雌性)指派到三个药剂组和一个介质对照组之一,在研究开始时(-1天)雄性年龄为2.5至4.4岁,雌性年龄为2.5至4.0岁,雄性体重为2.1至3.3kg,雌性体重为2.0至3.1kg。将5只动物/性别指派给每个药剂组并每日给药0(介质)、20、40或80mg/kg的口服药剂最长91天。由于在给药的前两周期间在给药80mg/kg/天的几只动物中发生癫痫发作,因此将高剂量降低至60mg/kg/天。在给药时间段结束时(第92天)将3只动物/性别/组按计划结束,将2只动物/性别/组指派到6周的不给药恢复阶段并在第134天结束。
评估动物的下列变化:临床征兆(笼子侧面观察和食物消耗[每天两次],以及给药后观察[每次给药当天给药后1-2小时]),体重(第-2和-1周,以及随后从第7天开始并在尸检之前每周),心电图,眼科检查,以及血压测量(研究前和第1、4和13周中),超声心动测量(第11周),临床病理指数包括血清化学、血液学和凝血(在给药开始之前一周内和接近第1、4、10和13周结束时;以及在接近恢复期结束时从剩余动物)和肌钙蛋白I(研究前;在第1、4和13周中在给药前和给药后2、4和24小时,以及在接近恢复期结束的单一时间点处)。用于尿液分析的尿样仅在尸检期间通过膀胱穿刺获得。用于尿液分析和尿液化学分析的尿样也在第-1、1、4和13周中以及在接近恢复期结束时从剩余动物,通过从特制不锈钢笼子盘引流获得。
在第1天和第4和14周期间,在各个时间点收集用于毒物动力学分析的血样。在最后一次给药后一天,将21只动物(3/性别/第1组,2只雄性/3只雌性/第2组,3只雄性/2只雌性/第3组和2只雄性/3只雌性/第4组)安乐死。将剩余的14只动物(来自于第1和2组各2只/性别,1只雄性/2只雌性/第3组和2只雄性/1只雌性/第4组)不进一步给药继续进行研究,并在最后一次给药后约6周安乐死。在结束时,对所有动物进行完全尸检,收集组织、防腐、处理,并由美国兽医病理学家学会(American College of Veterinary Pathologists)认证的兽医病理学家通过显微镜进行检查。
CEP-28122在食蟹猴中的4周口服毒性和毒物动力学研究,包括4周的恢复期
将CEP-28122(单甲磺酸盐单HCl盐)通过经口管饲法以0(介质对照)、3、10、20或40mg/kg/天的剂量水平给药于食蟹猴组(5只/性别/组)。在4周的治疗期后,3只动物/性别/组结束,2只动物/性别/组进入4周的无治疗恢复期。
在本研究中评估下列参数和终点:临床征兆(死亡/垂死检查,笼子侧面观察和食物消耗,以及给药后观察),体重,兽医身体检查,肺评估,眼科,心电图,血压和心率测量,临床病理参数(血液学、凝血、临床化学、尿液分析、尿液化学和肌钙蛋白I分析),毒物动力学参数,总体尸检发现,器官重量和组织病理学检查。
CEP-37440在Sprague-Dawley大鼠中的4周口服毒性和毒物动力学研究,包括4周的恢复期
将雄性和雌性大鼠(15/性别/组)指派到4个治疗组和一个介质对照组(pH调整过的反渗水),并按照下表中标出的给药作为二水合三盐酸盐的CEP-37440。动物通过经口管饲法给药。
a第1组仅接受介质对照(pH调整过的反渗水)。
b在最后一剂给药后经历4周恢复的被指定用于恢复期处死的毒性动物(在第1、2、3、4和5组中最多5只动物/性别)。
c表示成游离碱的剂量。
d药剂体积为10mL/kg。
毒性的评价是基于死亡率、临床观察、体重、体重变化、食物消耗、眼科检查和临床和解剖病理学。收集血样用于毒物动力学评估。
CEP-37440在食蟹猴中的4周口服毒性和毒物动力学研究,包括4周的恢复期
将雄性和雌性食蟹猴(Macaca fascicularis)指派到由5只猴/性别/组构成的4个组(3个治疗组和一个介质对照组)。在本研究中评估的剂量水平是0(pH调整过的反渗水)、2.5、7.5和20mg/kg/天。给药体积为5mL/kg。CEP-37440作为二水合三盐酸盐形式给药。在给药期完成后,将3只猴/性别/组安乐死,并对2只猴/性别/组继续进行另外4周的无治疗恢复期。
抗肿瘤效能研究–总体方案
将带有肿瘤的小鼠随机分配到不同治疗组中(8-10只小鼠/组),并以指定的剂量(表示成游离碱的mg/kg当量)和指定的给药频率(一日两次(bid)或一日四次(qid))经口给药介质(PEG-400)或配制在PEG-400中的试验化合物,使用每只小鼠100μL的给药体积。使用游标卡尺测量每个肿瘤的长度(L)和宽度(W),并且每2-3天测定小鼠体重。然后使用公式0.5236*L*W*(L+W)/2来计算肿瘤体积。使用Mann-Whitney秩和检验进行肿瘤体积和小鼠体重的统计分析。在每个剂量水平下最后一剂后2小时获得血浆和肿瘤样品,并通过LCMS/MS测量血浆和肿瘤裂解物中的化合物水平。
在小鼠中,在NPM-ALK阳性Sup-M2和Karpas-299 ALCL肿瘤异种移植物模型中的抗肿瘤效能
将PEG400中的CEP-37440一日一次(qd)或一日两次经口给药于含有NPM-ALK阳性Sup-M2ALCL肿瘤异种移植物的小鼠12天。还评估了CEP-37440(无定形HCl盐)在Scid小鼠中的第二种更具抗性的NPM-ALK阳性ALCL(Karpas-299)肿瘤异种移植物模型中的抗肿瘤效能。
使用口服给药在小鼠中的EML4-ALK阳性(NCI-H2228和NCI-H3122)NSCLC肿瘤异种移植物中的抗肿瘤活性
将人类肺癌细胞系NCI-H2228和NCI-H1650(ATCC,Manassas,VA)和NCI-H3122(由Dr.Giorgio Inghirami,Univ.of Torino,Italy善意提供)生长在增补有10%胎牛血清(FBS,Cat#SH3007003,Hyclone,Logan,UT)的RPMI-1640培养基中。NCI-H2228细胞带有EML4-ALK变体3a/b,NCI-H3122细胞含有EML4-ALK变体1,正如通过以前报道的荧光原位杂交和反转录PCR所确定的(Koivunen JP,Mermel C,Zejnullahu K,Murphy C,Lifshits E,Holmes AJ等,“EML4-ALK融合基因以及ALK激酶抑制剂在肺癌中的效能”(EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer),Clin Cancer Res 2008,14:4275-8)。
EML4-ALK阳性和EML4-ALK阴性NSCLC皮下肿瘤异种移植物在Scid小鼠中的产生:将雌性Scid/Beige小鼠(6-8周,Taconic,Hudson,NY)以5只/笼的密度维持在微型隔离器装置中和标准的实验室饮食(Teklad Labchow,Harlan Teklad,Madison,WI)下。将动物饲养在湿度和温度受控的条件下,并将光/暗周期设定为12小时的时间间隔。在实验操作之前,将小鼠隔离检疫至少一周。所有动物研究在Cephalon,Inc的学术动物护理和使用委员会(Institutional Animal Care and Use Committee)(IACUC)批准的方案(#03-023)下进行。简单来说,收集EML4-ALK阳性和阴性的NSCLC细胞,并以5x107/mL的密度重悬浮在RPMI-1640培养基中。使用23g针头(23G1,Cat#305145,Becton Dickinson,Franklin,NJ)将细胞悬液的等分试样(100μL)(5x106个细胞)皮下接种到每只小鼠的左胁。对小鼠进行监测,直至肿瘤异种移植物体积达到200-500mm3。
将带有肿瘤的小鼠随机分配到不同的治疗组中(8-10只小鼠/组),并以指定的剂量一日两次给药介质(PEG-400)或配制在PEG-400中的CEP-37440无定形HCl盐,使用每只小鼠100μL的给药体积。使用游标卡尺测量每个肿瘤的长度(L)和宽度(W),并且每2至3天测定小鼠体重。使用公式0.5236*L*W*(L+W)/2来计算肿瘤体积。肿瘤生长抑制百分率(%TGI)如下计算:(对照组在治疗结束时的肿瘤体积–治疗组在治疗结束时的肿瘤体积)/对照组在治疗结束时的肿瘤体积。肿瘤部分消退(PR)被定义为治疗组在治疗结束时的肿瘤体积小于治疗组在治疗开始时的肿瘤体积。肿瘤完全消退(CR)被定义为治疗组在治疗结束时的肿瘤体积小于治疗组在治疗开始时的肿瘤体积的5%。使用Mann-Whitney秩和检验进行肿瘤体积和小鼠体重的统计分析。在每个剂量水平下最后一剂后2小时获得血浆和肿瘤样品,并通过LCMS/MS测量血浆和肿瘤裂解物中的化合物水平。
在激素不依赖性前列腺癌、NSCL癌和HNSC癌的人类肿瘤异种移植物中的抗肿瘤效能研究
人类前列腺癌细胞系CWR22和PC3以及人类头颈部鳞状细胞癌细胞系Detroit 562从美国组织培养物保藏中心(American Tissue Culture Collection)(ATCC,Manassas,VA)获得。CWR22细胞培养在增补有10%胎牛血清(FBS,Cat#SH3007003,Hyclone Laboratory Inc,Logan,UT)的RPMI(ATCC,Cat#30-2001)中,PC3培养在增补有10%FBS的F12培养基(ATCC,Cat#30-2004)中,Detroit 562培养在增补有10%FBS的EMEM(ATCC,Cat#30-2003)中。人类非小细胞肺癌细胞系HCC-827和人类乳腺癌细胞系BT474也购自ATCC(Manassas,VA)并培养在含有10%FBS的RPMI(Cat#10-040,Mediatech Inc,Manassas,VA)中。兔磷酸-FAK(Tyr397)(Cat#3283)和FAK抗体(Cat#3285)购自Cell Signaling Technology(Beverly,MA)。
皮下人类肿瘤异种移植物在SCID/Beige或Nu/Nu小鼠中的产生。将雌性SCID/Beige(6-8周,Taconic,Hudson,NY)或Nu/Nu小鼠(6-8周,Charles River Laboratory,Wilmington,MA)以5只/笼的密度维持在微型隔离器装置中和标准的实验室饮食(Teklad Labchow,Harlan Teklad,Madison,WI)下。将动物饲养在湿度和温度受控的条件下,并将光/暗周期设定为12小时的时间间隔。在实验操作之前,将小鼠隔离检疫至少一周。实验得到Cephalon,Inc的学术动物护理和使用委员会(Institutional Animal Care and Use Committee)的批准(方案03-023)。简单来说,收集细胞并以5x107/mL的密度重悬浮在RPMI-培养基中,使用23g针头(23G1,Cat#305145,Becton Dickinson,Franklin,NJ)将细胞悬液的等分试样(100μL)(4x106或5x106个细胞)皮下接种到每只小鼠的左胁。然后每天对小鼠进行监测。
将带有肿瘤的小鼠随机分配到不同的治疗组中(8-10只小鼠/组),并以指定的剂量(表示成游离碱的mg/kg当量)和指定的给药频率口服给药介质(PEG-400)或配制在PEG-400中的CEP-37440,使用每只小鼠100μL的给药体积。使用游标卡尺测量每个肿瘤的长度(L)和宽度(W),并且每2-3天测定小鼠体重。然后使用公式0.5236*L*W*(L+W)/2来计算肿瘤体积。使用Mann-Whitney秩和检验进行肿瘤体积和小鼠体重的统计分析。在每个剂量水平下最后一剂后2小时获得血浆和肿瘤样品,并通过LCMS/MS测量血浆和肿瘤裂解物中的化合物水平。在研究结束时,通过将每个CEP-37440治疗组的肿瘤体积(TV)与介质治疗组的肿瘤体积进行比较,使用下列公式计算TGI值:[1-(化合物治疗组的最后一天的TV/介质治疗组的最后一天的TV)]*100。
VII.结果
CEP-37440和CEP-28122的生物学数据概述在表3中并在下文中提出和讨论。
表3.CEP-37440和CEP-28122的数据概述
·代谢稳定性/蛋白结合:(M)小鼠,(R)大鼠,(Mo)猴,(H)人类,(D)狗
·CEP-37440的小鼠和猴生物利用度实验使用CEP-37440无定形HCl盐来进行
这些数据表明,CEP-37440和CEP-28122两者都是强效ALK抑制剂。然而,与CEP-28122相比CEP-37440更加有利,因为CEP-37440是强效得多的FAK抑制剂,在人类血浆中具有降低的蛋白结合和提高的活性,具有提高的固有溶解性(便于吸收)、降低的亲脂性(较高的亲脂性已与提高的毒性相关联)、提高的代谢稳定性(促进在较低剂量下较高的血液浓度,这降低了对于身体的异型生物质负担——暴露于药物和代谢物越少,则对代谢系统例如肝脏的压力越小),由于P450抑制的降低而具有较低的药物-药物相互作用能力,尤其是对CYP3A4和CYP2C9来说(对同时给药的药物的正常代谢和清除的干扰降低),并具有提高的口服生物利用度和较低的体内清除率(在较低剂量下较高的血液浓度)。CEP-37440与CEP-28122相比的有利性质是令人吃惊和出人意料的。
CEP-28122在Sprague-Dawley大鼠中的13周口服毒性和毒物动力学研究,包括4周的恢复期
在研究期间没有药物相关的死亡并且对体重、食物消耗或心肌钙蛋白浓度没有药物相关的影响。对血液学参数的药物相关的影响限于雌性动物中极小的、无不利效应的红细胞、血红蛋白和血细胞比容的减少,以及在两种性别中极小的、无不利效应的血小板增加。还观察到其他统计学差异,并且由于变化的幅度或方向和/或缺少剂量依赖性而被认为是无意义的。
在雄性和雌性中,在150mg/kg/天下,在恢复时血小板保持轻度上升。对凝血参数的可能的药物相关的影响限于在接受150mg/kg/天的雄性中凝血酶原时间(PT)和部分促凝血酶原激酶时间(APTT)的延长以及在所有剂量水平的雌性中AP和APTT的缩短(雌性的值保持在预期范围内)。在恢复期结束时没有明显的有意义的变化。
临床化学参数的药物相关的变化限于接受≥75mg/kg/天的雄性中丙氨酸氨基转移酶(ALT)、总胆红素、天冬氨酸氨基转移酶(AST)、γ-谷氨酰转移酶(GGT)和山梨糖醇脱氢酶(SDH)的中度但不一致的增加。这种影响在13周给药期结束时比在4周的过渡样品收集时更显著,并且大多数可归因于在单一动物中的影响。在接受150mg/kg/天的雌性中AST和ALT也增加,但是这些影响的幅度一般远低于在雄性中观察到的。值得注意的是,在所有剂量水平下,雌性也表现出胆固醇的统计上显著、剂量依赖性和渐进的增加。在恢复期结束时,在给药期间接受75mg/kg/天的雄性个体中AST、ALT和SDH保持升高。在给药期间接受150mg/kg/天的雄性中,AST、ALT和SDH保持中度增加,而在给药期间接受150mg/kg/天的雌性中以较低幅度增加。
在接受≥75mg/kg/天的雄性和接受≥30mg/kg/天的雌性中,总蛋白和球蛋白剂量依赖性地增加。在接受≥75mg/kg/天的雌性中,白蛋白也统计上增加,但是值保持在预期范围内。钙的统计增加被认为是由于白蛋白和总蛋白增加而继发的。在结束时观察到其他零星的统计学差异,并且由于变化的幅度或方向和/或缺少剂量依赖性而被认为是无意义的。在恢复期结束时,给药对总蛋白、球蛋白、白蛋白或钙没有影响。
在结束时,在150mg/kg/天的雄性和雌性中,尿液体积增加并且比重降低。在结束或恢复时,在尿液分析参数中没有观察到其他显著变化。
在接受≥30mg/kg/天的雌性和接受≥75mg/kg/天的雄性中注意到肝重量的增加(相对于对照)(在75mg/kg/天下,雄性还具有相对于对照降低的胸腺重量)。在≥75mg/kg/天的剂量下,在两种性别中注意到肾、脾和肾上腺重量的增加。在≥75mg/kg/天的剂量下,心脏重量也中度增加。然而,由于在心脏中没有相关联的微观发现,因此这一发现的毒理学意义尚不清楚。在雄性的肾、心和脾中的影响持续到四周的恢复期结束。在雌性中,没有明显的持久的药物相关的影响。在13周给药期结束时,在接受150mg/kg/天的雄性和接受75mg/kg/天的雌性中在宏观上观察到肾脏的棕色或黑色变色。一个接受150mg/kg/天的雄性在恢复结束时,在肾脏上具有在微观上与肾小管色素相关的病灶。在13周给药期结束时,在接受≥30mg/kg/天的动物的肾、肾上腺、肝、肺、褐色脂肪组织、肠系膜淋巴结、脾和甲状腺中,药物相关的微观变化是明显的。在肾上腺、肝、肺、褐色脂肪组织、肠系膜淋巴结、脾和甲状腺中的发现,由于严重性低并且在4周的恢复期间有恢复的显著证据,因此不被认为是不利的。在接受≥75mg/kg/天的动物的肾脏中明显的渐进性神经病变,由于高的普及性以及在整个4周恢复期中的持久性,被认为是不利的。在恢复时,在几种组织中被推测是积累的药物的色素沉着,也是明显的,但是不被认为是不利的。
网状纤维的极小至轻度膨胀、尤其是在被膜下轴向皮层中的,在给药期间接受≥75mg/kg/天的恢复动物中是明显的。这一发现与这些动物中的临床白内障相符。然而,晶状体纤维的膨胀可能是用Davidson流体固定时由与乙酸相关的细胞膨胀造成的人为假象。被膜下皮层区域中晶状体纤维的膨胀,在被治疗的动物(包括上面列出的动物和没有临床白内障记录的其他被治疗动物)中,以及在较低程度上在本研究中的一些对照动物中,被注意到。此外,在几只以75和150mg/kg/天治疗的动物中,注意到在外周前皮质图案中晶状体纤维的膨胀,但是没有相应的检眼镜发现,这一发现被认为是由Davidson固定引起的继发性人为假象。由于这些原因,在本研究中观察到的晶状体纤维的膨胀被解释为人为假象,并且不被记录为显微镜发现。然而,被人为假象掩盖的真正生前改变的可能性不能被完全排除。
基于上面注意到的结果,受到在≥75mg/kg/天剂量下在肾脏中在整个4周的恢复期中持续的不利组织病理学影响的限制,本研究的无可注意到的副作用水平(No Observable Adverse Effect Level)(NOAEL)被认为是30mg/kg/天。
CEP-28122在食蟹猴中的13周口服毒性和毒物动力学研究,包括6周的恢复期
以20、40和80/60mg/kg的剂量水平每日通过鼻胃饲管给药CEP-28122连续91天,在所有剂量水平下引起几起与药物相关的不利事件,包括以80/60mg/kg给药的两只动物以及可能两只其他动物(一只以40mg/kg给药,另一只以20mg/kg给药)中的发病和死亡。还有另一只动物经历了早期尸检;但是这与CEP-28122给药无关。80mg/kg的给药在第9和/或10天引起2只动物癫痫发作,导致将这个组内所有动物中的剂量水平降低至60mg/kg。在食物消耗、体重、眼科检查、凝血参数、尿液分析参数和心率方面没有与CEP-28122相关的变化,在血压或肌钙蛋白I方面没有确定的与CEP-28122相关的变化。
经历早期尸检的动物的临床史一般可以分成两类:1)在几天/几周内健康状态逐渐下降的动物,以及2)在早期尸检当天给药后不久仍在临床上表现出耐受的动物。对于前一类中的动物来说,存在表明凝血病、急性期反应、严重脱水和/或肝毒性的临床病理变化,然而后一类中的动物大多数不存在临床病理变化。尽管在2类动物中存在快速发病(在给药的1-3分钟内)和死亡(在给药的15分钟内)的临床史表明药物灌输或吸入到肺中,但药剂给药到肺中不能根据组织病理学来鉴定。发病/死亡的快速发作的原因不能被确定,但是不能排除药物从肺的吸收。
在早期死亡动物中,在肺、肝、脾、肠系膜淋巴结和肾中鉴定到与药物相关的组织病理发现。在肺中,在所有药剂组中鉴定到与肺水肿相一致的总体和组织学发现,并且一般与如果存在的话存活至第92或134天的动物中的发现相比更严重。
在存活的动物中,存在着从所有剂量水平(20、40和80/60mg/kg)表现出的与CEP 28122相关的临床征兆,并包括:呕吐,活动减少,弓身外观和水状粪便。总的来说,这些临床征兆不是剂量依赖性的(除了水状粪便之外)。在研究的恢复期中没有检测到这些临床征兆。在给药80/60mg/kg的动物中存在着与CEP-28122相关的丙氨酸氨基转移酶(ALT)和天冬氨酸氨基转移酶(AST)的增加,表明了肝影响;然而,到恢复期结束时,这些值返回到基线。在肝中不存在确定的组织学关联性。
与CEP-28122相关的血液学参数的变化限于在给药80/60mg/kg的动物中淋巴细胞的减少,其在第28天开始并在研究的给药期间的整个剩余时间点中持续。到恢复期结束时淋巴细胞计数返回到基线。
在第92天,在肺、肝、脾、肠系膜淋巴结和肾中鉴定到与药物相关的组织病理学发现。在第92和134天,观察到与在早期死亡动物中鉴定到的类似的与肺水肿和更慢性的肺损伤相一致的组织学发现,它们不与剂量成比例,并且一般来说发生率和/或严重性与早期死亡动物相比更低。在给药40和80/60mg/kg CEP-28122的动物中通常鉴定到肝、脾和肠系膜淋巴结中巨噬细胞内的与剂量成比例的嗜曙红颗粒,包括在一只给药20mg/kg CEP-28122的动物中的极小变化。这一发现与一只高剂量动物的肝中肝细胞单细胞坏死相关,但是该高剂量动物在第134天时提高的发生率及其与嗜曙红颗粒的关联性,表明了与药物给药的关联性。在所有药物治疗组中鉴定到肾小管上皮细胞内与剂量成比例的棕色颗粒色素,它们与早期死亡动物中的极小至中度相反是极小至轻度的,并且与肾小管上皮细胞的退化/坏死无关。
在第92天,对于给药40和80/60mg/kg CEP-28122(肝和肾)和20、40和80/60mg/kg CEP-28122(肺)的雄性动物的肝、肺和肾来说,鉴定到与CEP-28122给药相关的器官重量和/或比率的增加。这些增加与肝中Kupffer细胞和肝细胞内的嗜曙红颗粒、肺中的肺水肿和肾中肾小管上皮细胞中棕色色素的积累相关。
在第134天,在肺、肝、脾、肠系膜和颌下淋巴结和肾中鉴定到与药物相关的组织病理学发现。在所有药剂组中,与终末期尸检动物相比,与肺水肿相一致的组织学变化类似地持续但严重性略低,并且同样不与剂量成比例。在一只高剂量动物中存在与更慢性的肺损伤相一致的极小变化。
通常在给药40和/或80/60mg/kg CEP-28122的动物中鉴定到肝、脾和肠系膜和颌下淋巴结中巨噬细胞内的嗜曙红颗粒并且是与剂量成比例的,其中除了脾之外,一个以上的组受到影响。肝脏中的这种变化一般来说比第92天时更严重。对于脾来说,对于80/60mg/kg组来说它的严重性与第92天相比降低,并且在20和40mg/kg组中在单一事件中具有可变的严重性。对于这些恢复组来说,肝和脾内嗜曙红颗粒区域内棕色颗粒色素的沉着程度与更早的时间点相比一般增加。与第92天相比,在给药40mg/kg CEP-28122的动物中肾的肾小管上皮细胞内与剂量成比例的棕色颗粒色素一般持续但严重性较低,在给药80/60mg/kg CEP-28122的动物中持续并且更加严重。在某些给药80/60mg/kg CEP-28122并具有最严重的色素积累的动物中,鉴定到肾小管上皮细胞的极小退化/坏死。
在第134天,在雌性动物中对于肝和肾来说鉴定到与CEP-28122给药相关的器官重量和/或比率的增加,其存在于给药40和/或80/60mg/kg CEP-28122的动物中,并且与终末期尸检动物中是相似相关的。
在本研究中肺水肿是重要的关注点。在许多情况下,在快速恶化和死亡之前肺水肿的发生是无症状的。此外,肺水肿的发生没有可感知的心血管组分,因此将这种发现分类为非心源性肺水肿。快速发作和缺少心血管影响使得在临床情况下诊断或预防肺水肿非常困难,使CEP-28122对于人类应用来说不安全。
CEP-28122在食蟹猴中的4周口服毒性和毒物动力学研究,包括4周的恢复期
CEP-28122以3、10、20和40mg/kg/天的剂量水平每天一次通过经口管饲法的4周给药没有引起任何发病或死亡。在所评估的任何剂量水平下,对食物消耗、体重、肺(通过听诊)、眼、ECG、血压、心率、血液学、凝血、尿液分析、尿液化学、肌钙蛋白I或总体病理学观察没有与CEP-28122相关的影响。
在≥10mg/kg/天的剂量水平下,出现与CEP-28122相关的组织学影响。这些发现由肺内空泡细胞(推定的肺泡上皮)的极小至轻度、多灶点的增加和极少的II型肺细胞增生(只在20mg/kg/天剂量水平下)构成。空泡细胞增加与来自于剂量水平≥10mg/kg/天的动物的肺重量和肺重量比的增加相关,但是与任何临床观察或通过听诊获得的肺的临床发现无关。在4周的恢复期后,在剂量水平≥20mg/kg/天的动物中肺内空泡细胞的增加仍然存在,尽管发生率和/或严重性较低。在10mg/kg/天的剂量水平下,出现这一发现的完全消解。在4周的恢复期后,没有从给药20mg/kg的动物检测到II型肺细胞增生,这也支持严重性的极小的全面降低。
可能与CEP-28122相关的影响包括在20和40mg/kg剂量水平下动物中的给药后呕吐以及甘油三酯的略微升高(只在40mg/kg剂量水平下)。在恢复期间没有检测到这些观察。在一只给药10mg/kg/天的雌性中在第29天时视网膜层的组织病理学改变,被认为是不确定的与CEP-28122相关的影响。在眼科检查中没有关联性。
这项在较低剂量下的4周研究证实,使用CEP-28122时不可能避免肺毒性。尤其值得关注的是发生肺损伤而没有先兆性征兆(听诊)这一事实。基于在4周和13周猴研究中肺毒性的出现和持久性得出结论,CEP-28122对于人类使用来说过于危险,并终止了开发尝试。
CEP-37440在Sprague-Dawley大鼠中的4周口服毒性和毒物动力学研究,包括4周的恢复期
在给药或恢复期间没有注意到与药物相关的临床观察。临床观察出现得相当不频繁,是暂时的,具有与对照相当的发生率,与濒死动物相关,与已知的饲管错误相关,或者发生在其死亡或处死被认为与药物无关的动物中;因此,临床观察不被认为是与药物相关的。
在给药期的所有时间间隔期间,给药30mg/kg/天的雄性和雌性与对照相比体重增加较少。在给药期的第1至28天期间,雄性比对照少增重31%。在给药期的第1至28天期间,雌性比对照少增重65%。在恢复期的所有时间间隔期间,给药30mg/kg/天的雄性和雌性与对照相比增加同样多或更多的体重。在恢复期的第1至28天期间,雄性比对照多增重24%。在恢复期的第1至28天期间,雌性比对照多增重29%。在给药期的所有时间间隔期间,给药30mg/kg/天的雄性和雌性比对照消耗更少食物。这些差异在雄性中在4至低于10%的范围内,在雌性中在12至低于24%的范围内。在恢复期间没有观察到对食物消耗的与药物相关的影响。在给药30mg/kg/天的雄性和雌性中,在终末期处死时观察到降低的平均终末期体重(分别为0.90x和0.79x),在雌性中是统计学显著的。
在给药或恢复期间没有注意到与药物相关的眼科发现。在高达10mg/kg/天下,在临床病理学试验结果中没有观察到与药物相关的影响。在30mg/kg/天下,观察到几例次要的临床病理影响,其程度为极小至轻度。这些发现都不被认为是不利的或毒理学上重要的。
30mg/kg/天下的药物血液学和凝血发现包括下列发现。
·在给药期的第29天的雌性中,轻度降低的红细胞质量(即红细胞计数、血红蛋白和红细胞比容)
·在给药期的第15和29天的雄性中和给药期的第15天的雌性中,轻度降低的网织红细胞绝对计数
·在给药期的第15和29天的雄性中,轻度降低的嗜中性粒细胞绝对计数
·在给药期的第29天的雌性中,轻度降低的嗜曙红细胞绝对计数
·在给药期的第15和29天的雄性中,极小升高的纤维蛋白原
血小板计数显得不受影响。红细胞质量、绝对网织红细胞、嗜中性粒细胞和嗜曙红细胞计数的降低可能反映出轻度的骨髓抑制/毒性,但是在骨髓中没有观察到相关的组织病理学发现。这些发现在恢复期结束时表现出可逆性。在恢复期结束时在给药30mg/kg/天的雌性中较高的平均红细胞容积,可能是由通常尺寸较大的较年轻红细胞的较高比例造成的。
在30mg/kg/天下,与药物相关的临床化学发现包括下列发现。
·在给药期的第29天的雌性中,轻度降低的白蛋白
·在给药期的第15和29天的雌性中,极小升高的球蛋白
·在给药期的第15和29天的雌性中,较低的白蛋白与球蛋白比率
·在给药期的第15和29天的雄性和雌性中,极小升高的胆固醇
·在给药期的第29天的雄性中,轻度升高的血清钙浓度
·在给药期的第29天的雄性和雌性中,极小降低的血清氯化物
较低的白蛋白和白蛋白与球蛋白比率和较高的球蛋白与炎症相符,并且可能与组织学上观察到的胸腔的慢性炎症相关。较高的胆固醇可能与在这些动物中观察到的减少的食物消耗和体重增加相关。较低的血清氯化物可能由较高的尿液排泄造成。较高的钙的机制是未知的。在任何剂量水平下,在肌钙蛋白I浓度中没有观察到与药物相关的影响。所有临床化学发现在恢复期结束时表现出可逆性。
在30mg/kg/天下,与药物相关的尿液化学发现包括下列发现。
·在给药期的第15和29天的雄性和雌性中,极小升高的尿液氯化物排泄
·在给药期的第15和29天的雄性和雌性中,极小升高的尿液氯化物清除分数
较低的血清氯化合物可能与给药30mg/kg/天的动物的尿液中较高的总排泄和较高的氯化物清除分数相关。在这些动物的肾脏中没有观察到相关的组织病理学发现。在恢复期结束时,所有尿液化学发现表现出可逆性。在任何剂量水平下,在尿液分析试验结果中没有观察到明显的影响。
在其他临床病理学试验结果中统计学显著的或以其他方式可注意到的差异被认为是偶然的,因为它们通常幅度小、缺乏与剂量的关系或者随时间并且在性别之间不一致。
在给药期间,在毒性动物中发生7例计划外死亡。所有计划外死亡都不归因于药物。一只对照雄性和一只给药30mg/kg/天的雌性在收集血液后不久死亡;由于缺乏表明与药物相关的影响的临床或解剖病理学发现,它们被认为是意外死亡。三只动物具有与饲管相关的损伤相一致的宏观和/或微观发现,包括一只给药1mg/kg/天的雌性在第25天在濒死状态下被处死,一只给药3mg/kg/天的雌性被发现在第9天死亡,一只给药10mg/kg/天的雌性在给药期的第25天在濒死状态下被处死。对于在第10天被发现死亡的一只给药30mg/kg/天的雄性和在给药期第15天在濒死状态下被处死的一只给药10mg/kg/天的雌性来说,死亡原因不清楚。所有其他给药期和所有恢复期毒性动物存活到它们的计划处死。
在终末期处死时,在给药30mg/kg/天的雄性的肝脏和给药10mg/kg/天的雄性的前列腺中,发生平均器官重量(对于终末期体重进行调整)的统计学显著的增加。由于这些改变缺乏微观关联物、性别之间的一致性(仅仅肝脏)和/或剂量响应性的证据,它们不被认为是与药物相关的。在给药30mg/kg/天的雄性(0.86x)和给药10mg/kg/天(0.85x)或30mg/kg/天(0.79x)的雌性中,调整过的平均胸腺重量降低超过10%,并且呈剂量依赖性方式。由于缺少统计学显著性或微观关联物,在这些组中胸腺重量降低与药物的关系是不确定的。在终末期处死时所有其他器官的重量变化和恢复期处死时所有器官的重量变化,可能是由正常的生物变差造成的,并且不被认为是药物的直接影响。
在给药30mg/kg/天的雌性中,在终末期处死时观察到心脏中的粘连和肺中的粘连或物质的宏观发现,并且在一只给药30mg/kg/天的雌性中在恢复期处死时观察到心脏中的粘连。这些宏观发现与心脏和肺中纤维化和/或慢性炎症的微观发现相关,并且被认为是与药物相关的。所有其他宏观发现被认为是自发的、偶然的或与意外死亡相关,并且不归因于药物。
在终末期处死时,在给药30mg/kg/天的雌性的心脏和肺的浆膜表面上以及胸腺周边的结缔组织中存在与胸腔的慢性炎症相符的微观发现。在给药30mg/kg/天的5/10雌性中,观察到心脏的浆膜/心外膜表面和肺的胸膜/胸膜下表面的慢性炎症和/或纤维化。所述发现是多灶点到沿着心脏和肺的浆膜表面弥散的,并且从具有很少炎性细胞的纤维性增厚到更严重的慢性炎症不一而足。在给药30mg/kg/天的4/10雌性中,在围绕胸腺的松散的纤维血管结缔组织中也观察到慢性至慢性-活跃的炎症。在恢复期处死时,在给药30mg/kg/天的2/5雌性中观察到心脏和肺的浆膜表面的慢性炎症和/或纤维化。在恢复期处死时的发现,以多灶点到弥散的具有很少炎症的浆膜表面纤维性增厚为特征,表明部分缓解。由于在给药30mg/kg/天的总共7/15雌性中在计划处死时观察到胸腔中慢性炎症的发现,并且由于缺乏这些发现的另一种病因的宏观或微观证据,因此给药30mg/kg/天的雌性的心脏和肺的浆膜表面上和胸腺周边的纤维脂肪组织中的纤维化和/或炎症,被认为最可能是与药物相关的,并且可能是腹膜和心周积液继发的浆膜炎症的结果。
在给药30mg/kg/天的雌性中,在终末期处死时观察到肺中肺泡巨噬细胞发生率的小量增加,并且被认为最可能与药物相关。在恢复期处死时没有观察到肺泡巨噬细胞发生率的增加,表明这种发现是可逆的。在计划和计划外死亡时的所有其他微观发现被认为是自发的、偶然的或与意外死亡相关,并且不可归因于药物。
在以前进行的使用CEP-37440的10天剂量范围寻找研究中,每天通过饲管以10、30或60mg/kg/天向大鼠给药药物,在10mg/kg/天下是耐受良好的。在给药≥30mg/kg/天的动物中,观察到体重的与药物相关的不利发现与骨髓(细胞过少)、脾、胸腺和淋巴结(淋巴细胞减少)和肺(肺泡巨噬细胞的增加)的解剖病理学发现的组合。在给药10mg/kg/天的动物中没有观察到与药物相关的变化。在当前的研究中,在给药30mg/kg/天的雄性和雌性中观察到不利的并且与药物相关的发现。这些发现包括雄性和雌性中较低的体重增加,雄性和雌性中较低的食物消耗,以及雌性中与胸腔的慢性炎症相符的微观发现。
在最高10mg/kg/天的临床病理学试验结果中没有观察到与药物相关的影响。在30mg/kg/天下观察到几例次要的临床病理学影响,其程度为极小至轻度。这些发现都不被认为是不利、毒理学上重要或明确地与其他不利的生命或解剖病理学发现相关。
红细胞质量、网织红细胞、嗜中性粒细胞和嗜曙红细胞绝对计数的降低,可能反映出轻度的骨髓抑制/毒性,但是在骨髓中没有观察到相关的组织病理学发现。在恢复期结束时,这些发现表现出可逆性。在恢复期结束时在给药30mg/kg/天的雌性中较高的平均红细胞容积,可能是由通常尺寸较大的较年轻红细胞的较高比例造成的。
较低的白蛋白和白蛋白与球蛋白比率和较高的球蛋白与炎症相符,并且可能与组织学上观察到的胸腔的慢性炎症相关。较高的胆固醇可能与在这些动物中观察到的减少的食物消耗和体重增加相关。较低的血清氯化物可能由较高的尿液排泄造成。较高的钙的机制是未知的。在任何剂量水平下,在肌钙蛋白I浓度中没有观察到与药物相关的影响。所有临床化学发现在恢复期结束时表现出可逆性。
较低的血清氯化合物可能与给药30mg/kg/天的动物的尿液中较高的总排泄和较高的氯化物清除分数相关。在这些动物的肾脏中没有观察到相关的组织病理学发现,并且在恢复期结束时,所有尿液化学发现表现出可逆性。
结论是,以≤10mg/kg/天的剂量向大鼠口服给药CEP-37440 28天,在临床上良好地耐受。在给药30mg/kg/天的雌性中存在与胸腔的慢性炎症相符的微观发现。在给药30mg/kg/天的雄性和雌性中存在体重变化和食物消耗的降低。基于这些发现,在本研究中无可注意到的副作用水平(NOAEL)是10mg/kg/天。
CEP-37440在食蟹猴中的4周口服毒性和毒物动力学研究,包括4周的恢复期
所有动物存活至它们的计划处死。
在给药或恢复期间没有注意到与药物相关的临床观察。临床观察表现得相当不频繁,是暂时的,或者与对照发生率相当;因此,它们被认为是与药物无关的。
在给药或恢复期间,没有注意到对体重或体重增加的与药物相关的影响。没有注意到与药物相关的异常眼科发现。此外,在血压测量期间没有注意到与药物相关的观察。
在给药2.5、7.5或20.0mg CEP-37440/kg体重/天(mg/kg/天)的动物中,在给药期的第3和27天或恢复期的第28天,没有观察到PR间期、QRS持续时间、QT间期、校正的QT(QTc)间期、RR间期或心率的与药物相关的变化。在ECG的定性评估期间,没有观察到可归因于CEP-37440的节律异常或定性ECG变化。
最高20.0mg/kg/天的CEP-37440给药,在给药或恢复期结束时对临床病理学试验结果没有影响。
几只个体动物在给药期间具有与炎症相符的临床病理学发现(包括白细胞和嗜中性粒细胞绝对计数、纤维蛋白原和C反应蛋白的轻微至可注意到的增加),但是这些动物通常散布在所有组、包括对照中。就此而言,这些发现被认为是与药物无关的,因为它们缺少剂量相关模式,通常随时间而不一致(特别是白细胞计数),并且包括一些对照动物。
在终末期处死时,终末期体重或器官重量没有统计上显著的变化。在计划的终末期或恢复期处死时出现的所有器官重量变化,被归因于正常的生物变差,并且被认为是偶然且与药物无关的。
在终末期或恢复期处死时,不存在清楚的与药物相关的宏观发现。在终末期处死时,给药7.5mg/kg/天的所有(3/3)雄性和给药20.0mg/kg/天的2/3雌性,在胃的粘膜表面上表现出一个至几个红色至暗红色的灶点。在一只给药20.0mg/kg/天的雄性中,胃的浆膜表现出几个红色区域。微观关联物是粘膜和/或粘膜下层/肌膜内的灶性出血,并且在肌膜内存在或不存在平滑肌变性。在一只对照雌性中存在类似发现,表明这些发现是偶然的并且与药物无关。
在终末期或恢复期处死时,不存在清楚的与药物相关的微观发现。在一些给药CEP-37440的动物中胃的粘膜或浆膜中的宏观发现,通常与粘膜和/或粘膜下层/肌膜内的灶性出血并且在肌膜内存在或不存在平滑肌变性的微观发现相关联。然而,由于在给药对照物质的雌性的胃中存在类似和更严重的微观发现,以及胃平滑肌变性在食蟹猴中是已知的背景发现这一事实,在本研究中宏观和微观发现与CEP-37440给药的关联性是不确定的。在一只或多只动物的胃中存在其他微观发现,但是它们与药物的关联性是不确定的,这是因为它们的严重性和/或发生率低,缺乏明显的剂量响应,或者同时存在于对照动物中。
在终末期和恢复期处死时所有其余的微观发现,包括在两只给药20.0mg/kg/天的雌性的肺中肺泡巨噬细胞的极小的灶性浸润,被归因于正常的生物变差并被认为是偶然的。
结论是,以最高20mg/kg/天的剂量向食蟹猴口服给药CEP-37440连续28天是耐受良好的,并且在任何剂量水平下没有观察到与药物相关的影响。重要的是,没有任何肺水肿的迹象,并且CEP-37440被确定可安全用于人类试验。CEP-37440与CEP-28122相比优越的安全性情况,尤其是缺乏肺毒性,是令人吃惊和出人意料的。
在小鼠中的NPM-ALK阳性Sup-M2和Karpas-299 ALCL肿瘤异种移植物模型中的抗肿瘤效能
在以10mg/kg或更低剂量一日两次用CEP-37440治疗12天后,没有观察到显著的抗肿瘤活性;在以30mg/kg一日一次用CEP-37440治疗12天后,观察到部分的肿瘤减退;在以30mg/kg一日两次或55mg/kg一日一次用CEP-37440治疗12天后,观察到完全或接近完全的肿瘤减退(图4)。在所有剂量方案下,CEP-37440给药被良好地耐受,没有明显的毒性并且没有显著的与化合物相关的小鼠体重减轻(图5)。在最终给药后2h收集的血浆和肿瘤裂解物中,发现与剂量相关的水平的CEP-37440(图6)。注意到在30mg/kg一日两次和55mg/kg一日一次给药水平下的肿瘤中,没有可观察到的CEP-37440,因为那些动物没有肿瘤——肿瘤完全减退。血浆中的CEP-37440水平比PK/PD研究中单次口服药剂后2h时的水平高约2-3倍,肿瘤中的CEP-37440水平比其高10倍以上,表明使用10和30mg/kg下的一日两次或一日一次口服给药方案时,一些化合物蓄积在血浆和肿瘤中。
在Karpas-299肿瘤异种移植物中,在30mg/kg一日一次下观察到显著的抗肿瘤活性,并且在以30mg/kg一日两次或55mg/kg一日一次治疗12天后,观察到完全或接近完全的肿瘤减退(图7)。在所述给药方案下,CEP-37440给药耐受良好,没有明显的毒性,并且没有显著的体重减轻(图8)。在最终给药后2h收集的血浆和肿瘤裂解物中,观察到与剂量相关的水平的CEP-37440(图9)。注意到在50mg/kg一日一次给药水平下的肿瘤中,没有可观察到的CEP-37440,因为那些动物没有肿瘤——肿瘤完全减退。
在小鼠中的EML4-ALK阳性(NCI-H2228和NCI-H3122)NSCLC肿瘤异种移植物模型中口服给药的抗肿瘤活性
对于NCI-H2228肿瘤异种移植物模型来说,使用CEP-37440(HCl盐)以30mg/kg一日一次和一日两次以及55mg/kg一日一次口服给药治疗12天,引起肿瘤减退(图10)。对于NCI-H3122肿瘤异种移植物模型来说,使用CEP-37440(HCl盐)以30mg/kg一日两次或55mg/kg一日一次口服给药治疗12天,引起肿瘤停滞和部分减退(图11)。在NCI-H2228肿瘤异种移植物中观察到的提高的抗肿瘤效能可能是由CEP-37440的较高肿瘤分布造成的(图12和13)。除了在带有NCI-H3122肿瘤的小鼠中在30mg/kg一日两次剂量下(图15)之外,这些带有肿瘤的小鼠中的治疗是耐受良好的,没有明显的毒性或与化合物相关的体重减轻(图14和15)。
带有NCI-H2228肿瘤的小鼠以55mg/kg一日一次口服给药继续治疗4周,在100%的动物中提供了持续的完全肿瘤减退(图16)。继续治疗耐受良好,没有明显的毒性并且没有显著的体重减轻(图17)。在治疗停止后40天,在100%的小鼠中观察到持续的肿瘤减退,在任何小鼠中没有肿瘤重新出现(图16)。
这是重要的,因为它表明在使用CEP-37440治疗约6周后,肿瘤被完全根除并且小鼠被有效地“治愈”。
在激素不依赖性前列腺癌、NSCL癌和HNSC癌的人类肿瘤异种移植物中的抗肿瘤效能研究
在已建立的FAK阳性PC-3前列腺肿瘤异种移植物中,在36天的时间段中给药CEP-37440引起55%的肿瘤生长抑制(TGI)和10%的完全肿瘤减退发生率,所述情况与这一模型中等同剂量的PF-562271的情况(69%的TGI和25%的部分肿瘤减退发生率)类似(图18)。所有给药方案耐受良好,没有观察到明显毒性或显著的体重减轻。
非小细胞肺(NSCL)癌
通过28天的研究,CEP-37440被证实具有剂量相关的抗肿瘤效能,在55mg/kg一日两次下具有80%的TGI和60%的肿瘤减退发生率(30%完全,30%部分),并且在30mg/kg一日两次下具有显著效能(60%的TGI和部分肿瘤减退的迹象)(图19)。在55mg/kg一日两次下使用PF-562271观察到显著的抗肿瘤效能(66%TGI),但是在第23天开始观察到中度的肿瘤生长反弹。CEP-37440和PF-562271两者的给药耐受良好,没有观察到明显的毒性或显著的体重减轻(图20)。值得注意的是,使用CEP-37440获得的显著效能不是在该EML4-ALK阴性肿瘤异种移植物模型中抑制EGF-R磷酸化(活化)的结果。
头颈部鳞状细胞癌(HNSCC)
在带有已建立的Detroit 562HNSCC异种移植物的SCID小鼠中,CEP-37440和PF-562271对FAK激活的抑制表现出明显的肿瘤药效学效果,并且对总FAK表达水平没有影响(图21)。在28天的时间段中,CEP-37440和PF-562271引起肿瘤停滞,以及20%(CEP-37440,30mg/kg一日两次)和30%(CEP-37440和PF-562271,55mg/kg一日两次)的部分肿瘤减退发生率。使用CEP-37440(两种剂量)观察到的效能幅度与使用55mg/kg一日两次的PF-562271所观察到的(Roberts等,2008)相当。给药方案耐受良好,没有观察到发病或死亡。
这些研究是重要的,因为它们显示,CEP-37440在激素不依赖性前列腺癌、NSCL癌和HNSCC的已建立的异种移植物模型中表现出显著的FAK药效学抑制和ALK不依赖性抗肿瘤效能——包括客观肿瘤响应。
应该理解,本文中描述的实施例和实施方式仅仅是出于说明的目的,本领域的技术人员可以根据它们提出各种修改和改变,所述修改和改变将包含在本申请的精神和范围以及权利要求书的范围之内。本文中引用的所有出版物、专利和专利申请,以其全部内容为所有目的通过参考并入本文。
- 还没有人留言评论。精彩留言会获得点赞!