一种琥珀酸去甲文拉法辛一水合物新晶型及制备方法与流程
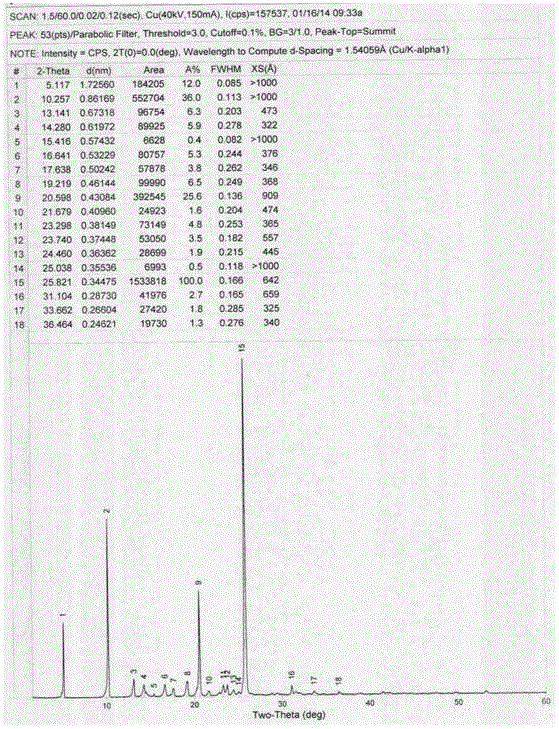
本发明涉及一种抗抑郁药物的新晶型及其制备方法,具体涉及一种琥珀酸去甲文拉法辛一水合物新晶型及其制备方法。
背景技术:
:琥珀酸去甲文拉法辛,化学名为4‐[2‐(二甲基氨基)‐1‐(1‐羟基环己基)乙基]苯酚琥珀酸盐,其一水合物结构式如下。琥珀酸去甲文拉法辛是文拉法辛的主要活性代谢物的琥珀酸盐,具有双重作用机制,为选择性神经系统5‐羟色胺(5‐HT)及去甲肾上腺素(NE)再摄取抑制剂(SNRI),能同时阻滞突触前5‐HT和NE的再摄取,导致这两种神经递质水平的持续升高,并轻度抑制多巴胺(DA)的摄取而发挥抗抑郁作用。本品对很多受体的亲和力都很小,如体外的毒蕈碱受体、胆碱能受体、H1组胺能神经受体和α1肾上腺素能受体。琥珀酸去甲文拉法辛(商品名Pristiq)由美国惠氏公司研制开发,是美国FDA批准的治疗重性抑郁障碍(MDD)的首个SNRI,是目前全球处方用药最广泛的抗抑郁药。惠氏公司在中国专利CN1501909A中公开了琥珀酸去甲文拉法辛的4种晶型。其中晶型Ⅰ和Ⅱ为琥珀酸去甲文拉法辛一水合物的两种晶型,Ⅲ型为介于半水合物和一水合物之间的琥珀酸去甲文拉法辛水合物的晶型,Ⅳ型为琥珀酸去甲文拉法辛无水物的晶型。晶型Ⅰ的峰位和相对强度数据如下:度2θ±0.2°2θI/I110.201714.911220.561822.131123.711324.601425.79100制备晶型Ⅰ有两种方法,一是将O‐去甲文拉法辛的游离碱与琥珀酸溶解于丙酮水溶液中,然后将溶液缓慢冷却(例如,3小时或更长);二是利用Ⅰ、Ⅱ和Ⅲ型或其混合物与丙酮、乙腈、乙腈‐水混合物,或乙醇‐甲苯混合物的浆液来制备晶型Ⅰ。晶型Ⅱ的峰位和相对强度数据如下:度2θ±0.2°2θI/I110.252213.181414.041014.353514.661816.685217.672919.242920.381620.562523.412423.781624.571325.131025.8010031.7814Ⅱ型的制备方法有多种:一是将溶解于丙酮的Ⅰ型旋转蒸发制备Ⅱ型,二是缓慢冷却Ⅰ型的饱和丙酮溶液或95:5乙醇‐水溶液(冷却过程十分缓慢),三是从乙醇/乙烷的溶剂/抗溶剂的混合物中沉淀Ⅰ型,四是从水中缓慢蒸发Ⅰ型,五是由乙腈或乙醇/乙烷或乙醇/氯仿母液中快速蒸发Ⅰ型,六是快速冷却琥珀酸去甲文拉法辛的水溶液或水/丙酮溶液,七是将无定型的琥珀酸去甲文拉法辛置于75%或更大的相对湿度下。US20090234020的实施例5也公开了去甲文拉法辛琥珀酸盐一水合物的制备方法,将去 甲文拉法辛50g(1eq)溶解于250ml(5v/w)异丙醇中,加热至回流(83℃),琥珀酸24.65g(1.07eq)溶于100ml(2v/w)水中,加入反应体系,回流1h,5h内降温至25℃,搅拌过夜。收率90%。为使描述简便,本发明中称其为晶型M。技术实现要素:本发明提供了一种新的琥珀酸去甲文拉法辛一水合物晶型,即晶型Ⅵ。值得注意的是,获得一种新的晶型并非本发明的唯一目的。本发明的晶型Ⅵ以药用低密度聚乙烯袋封装后在40℃±2℃/75%±5%RH条件下,经加速6个月以及在25℃±2℃/60%±5%RH条件下,各项指标无明显变化,均符合质量标准要求,稳定性良好。并且相比于晶型Ⅰ、Ⅱ和M,晶型Ⅵ制片收率最优,所制得的缓释片释放曲线更接近市售制剂(实验及结果详见“具体实施方式”部分)。本发明提供的琥珀酸去甲文拉法辛晶型Ⅵ,在以下位置上有特征峰:5.14°、10.26°、13.16°、14.28°、16.67°、19.14°、20.60°、23.30°和25.86°。进一步地,本发明提供的琥珀酸去甲文拉法辛晶型Ⅵ在以下位置上有特征峰:5.14°、10.26°、13.16°、14.28°、15.42°、16.67°、17.60°、19.14°、20.60°、21.66°、23.30°、23.72°、24.48°、25.04°、25.86°、31.16°、33.64°和36.54°。更进一步地,本发明提供的琥珀酸去甲文拉法辛晶型Ⅵ,其具有与附图1基本相同的X‐射线粉末衍射图谱。由于测量条件的不同,X‐射线粉末衍射图上各峰的2θ角和相对强度会有所变动,一般2θ角变化在±0.2以内,相对强度在±0.2%以内认为是合理误差。本发明提供的琥珀酸去甲文拉法辛晶型Ⅵ差示扫描量热分析(DSC)如附图3所示,显示三个吸收峰,分别约为103℃、119℃和234℃。结合热重分析(TGA),在80‐155℃范围失重约4.5%(重量比)。琥珀酸去甲文拉法辛一水合物理论含水量为4.5%(重量比),因此琥珀酸去甲文拉法辛晶型Ⅵ为一水合物。在167‐270℃区间,失重约61.5%,应为分解失重。表1显示了本发明的琥珀酸去甲文拉法辛一水合物晶型Ⅵ的X‐射线粉末衍射数据(仅列出相对强度>5%的衍射峰数据)。表1.琥珀酸去甲文拉法辛一水合物晶型Ⅵ的X‐射线粉末衍射数据度2θ±0.2°2θI/I15.141610.263713.16814.28916.67619.141020.602423.30525.86100表2.琥珀酸去甲文拉法辛一水合物晶型Ⅰ的X‐射线粉末衍射数据度2θ±0.2°2θI/I110.201714.911220.561822.131123.711324.601425.79100对比晶型Ⅰ和晶型Ⅵ的X‐射线粉末衍射数据可以明显看出,晶型Ⅵ与晶型Ⅰ有显著区别,晶型Ⅰ在22.13°、23.71°和24.60°位置有特征峰,而晶型Ⅵ在这些位置没有明显特征峰;晶型Ⅵ在5.14°、13.16°、16.67°、19.14°和23.30°有特征峰,而晶型Ⅰ在这些位置没有明显特征峰。因此,晶型Ⅵ与晶型Ⅰ是两种不同的晶型。表3.琥珀酸去甲文拉法辛一水合物晶型Ⅱ的X‐射线粉末衍射数据度2θ±0.2°2θI/I110.252213.181414.041014.353514.661816.685217.672919.242920.381620.562523.412423.781624.571325.131025.8010031.7814对比晶型Ⅱ和晶型Ⅵ的X‐射线粉末衍射数据可以明显看出,晶型Ⅵ与晶型Ⅱ也有显著区别,晶型Ⅱ在14.04°、14.66°、17.67°、20.38°、23.78°、24.57°、25.13°和31.78°位置有特征峰,而晶型Ⅵ在这些位置没有明显特征峰。因此,晶型Ⅵ与晶型Ⅱ是两种不同的晶型。本发明的发明人按照专利US20090234020方法制备的琥珀酸去甲文拉法辛一水合物晶型M(方法详见对比例3),其X‐射线粉末衍射数据如表4所示。表4.琥珀酸去甲文拉法辛一水合物M晶型X‐射线粉末衍射数据度2θ±0.2°2θI/I15.222710.383311.961815.082216.66819.04520.764222.38823.98924.82926.02100与晶型Ⅵ的X‐射线粉末衍射数据可以明显看出,二者有显著区别。专利US20090234020方法制备的琥珀酸去甲文拉法辛在11.96°、15.08°和22.38°位置有特征峰,而晶型Ⅵ在这些位置没有明显特征峰。晶型Ⅵ在13.16°和14.28°有特征峰,而专利US20090234020方法制备的琥珀酸去甲文拉法辛在这些位置没有明显特征峰。因此,上述晶型是两种不同的晶型。将本发明实施例提供的琥珀酸去甲文拉法辛一水合物晶型Ⅵ,以药用低密度聚乙烯袋封装后在40℃±2℃/75%±5%RH条件下,经加速6个月以及在25℃±2℃/60%±5%RH条件下,各项指标无明显变化,均符合质量标准要求,稳定性良好(详见实施例5和6)。将本发明实施例提供的琥珀酸去甲文拉法辛一水合物晶型Ⅵ,以及按照CN100567253C公开的方法制备的晶型Ⅰ和晶型Ⅱ,按照US20090234020公开的方法制备的琥珀酸去甲文拉法辛(详见对比例1、2和3),按相同制剂配方及工艺制备成同规格片剂,晶型Ⅵ制片收率最优(详见实施例7),体外溶出试验,晶型Ⅵ缓释片释放曲线更接近市售制剂(详见实施例8)。本发明的另一目的在于提供所述琥珀酸去甲文拉法辛晶型一水合物Ⅵ的制备方法,该方法可直接得到本发明所述琥珀酸去甲文拉法辛晶型一水合物Ⅵ,纯度99.5%以上,收率高于95%,且工艺简单,条件温和,极大地缩短了反应时间,提高了生产效率。本发明提供的琥珀酸去甲文拉法辛晶型一水合物Ⅵ的制备方法包括如下步骤:①将琥珀酸和去甲文拉法辛溶于或悬浮于异丙醇与水组成的混合溶剂中,升温至回流温度,保温搅拌0.5‐1小时,其中异丙醇与水的体积比为7:2‐20:1;②调温至析晶温度,搅拌析晶4‐5小时;③分离;④干燥,得琥珀酸去甲文拉法辛一水合物晶型Ⅵ。其中,步骤①去甲文拉法辛与琥珀酸的投料摩尔比例为1:1.0‐1:1.2,优选1:1.05。其中,步骤①中去甲文拉法辛与混合溶剂的质量体积比例为1:9‐1:12,优选1:11.55。“质量体积比例”是指去甲文拉法辛的重量(g)与混合溶剂的体积(mL)之比。其中,异丙醇与水的体积比为20:1。其中,步骤②析晶温度为20‐50℃。进一步地,步骤②析晶温度为20‐30℃。虽然US20090234020的实施例5公开了以异丙醇‐水(二者比例5:2)作为溶剂系统,但其析晶时需要搅拌过夜,且收率仅为90%。然而本发明的发明人在大量的实验中发现,以异丙醇‐水混合溶剂作为结晶溶剂并限定二 者比例为7:2‐20:1,特别是异丙醇与水的体积比为20:1时,得到了一种琥珀酸去甲文拉法辛一水合物的新晶型,即本发明定义的晶型Ⅵ。而其析晶过程仅仅需要4‐5小时,极大地缩短了反应时间,提高了生产效率;并且收率均高于95%,对于工业化生产意义重大。定义:术语“晶型”指琥珀酸去甲文拉法辛或其水合物的晶体空间结构取向性变化。可通过X‐射线粉末衍射、热重分析(TGA)、差示扫描量热分析(DSC)、测定熔点或其它方法确定。术语“约”指在给出数值或范围的10%,优选5%,更优选1%以内。术语“约”也可以指本领域普通技术人员可接受的标准误差范围内。附图说明附图1本发明琥珀酸去文拉法辛一水合物晶型Ⅵ的X‐射线粉末衍射数据及图谱。附图2本发明琥珀酸去文拉法辛一水合物晶型Ⅵ加速稳定性试验X‐射线粉末衍射数据及图谱。附图3本发明琥珀酸去文拉法辛一水合物晶型Ⅵ长期稳定性试验X‐射线粉末衍射数据及图谱。附图4本发明琥珀酸去文拉法辛一水合物晶型Ⅵ的差示扫描量热分析和热重分析(DSC‐TGA)图谱。具体实施方式以下实施例是对本发明的具体说明,不应对本发明的范围构成限制。实施例1:琥珀酸去甲文拉法辛晶型Ⅵ的制备将去甲文拉法辛263g(1mol,1.0eq)和琥珀酸124g(1.05mol,1.05eq)加入反应瓶中。加入异丙醇1.84L(7v/w)、纯化水526mL(2v/w)。搅拌升温至回流,保温搅拌1h,停止加热。自然降温至0~10℃,搅拌析晶4.5h,抽滤,滤饼用少量异丙醇淋洗1次,将滤饼转移至真空干燥箱,控制温度40~50℃真空干燥6h,得白色固体385g,收率96.5%,纯度99.83%。将所得结晶样品进行粉末X‐射线粉末衍射,所得图谱及数据见附图1。实施例2:琥珀酸去甲文拉法辛晶型Ⅵ的制备将去甲文拉法辛263g(1mol,1.0eq)和琥珀酸124g(1.05mol,1.05eq)加入反应瓶中。加入异丙醇2.10L(8v/w)、纯化水526mL(2v/w)。搅拌升温至回流,保温搅拌1h,停止加热。自然降温至20~30℃,搅拌析晶4.5h,抽滤,滤饼用少量异丙醇淋洗1次,将滤饼转移至真空 干燥箱,控制温度40~50℃真空干燥6h,得白色固体383g,收率96.0%,纯度99.81%。其X‐射线粉末衍射图谱与附图1一致。实施例3:琥珀酸去甲文拉法辛晶型Ⅵ的制备将去甲文拉法辛263g(1mol,1.0eq)和琥珀酸124g(1.05mol,1.05eq)加入反应瓶中。加入异丙醇2.90L(11v/w)、纯化水263(1v/w)mL。搅拌升温至回流,保温搅拌0.5h,停止加热。自然降温至30~40℃,搅拌析晶4.5h,抽滤,滤饼用少量异丙醇淋洗1次,将滤饼转移至真空干燥箱,控制温度40~50℃真空干燥6h,得白色固体380g,收率95.2%,纯度99.79%。其X‐射线粉末衍射图谱与附图1一致。实施例4:琥珀酸去甲文拉法辛晶型Ⅵ的制备将去甲文拉法辛263g(1mol,1.0eq)和琥珀酸124g(1.05mol,1.05eq)加入反应瓶中。加入异丙醇2.90L(11v/w)、纯化水145mL(0.55v/w)。搅拌升温至回流,保温搅拌0.5h,停止加热。自然降温至20~30℃,搅拌析晶4.5h,抽滤,滤饼用少量异丙醇淋洗1次,将滤饼转移至真空干燥箱,控制温度40~50℃真空干燥6h,得白色固体385g,收率96.5%,纯度99.83%。其X‐射线粉末衍射图谱与附图1一致。实施例5长期稳定性试验将实施例4提供的琥珀酸去甲文拉法辛一水合物晶型Ⅵ,以药用低密度聚乙烯袋封装后在25℃±2℃/60%±5%RH条件下,进行6个月长期稳定性试验,结果如下:表5.长期稳定性试验结果实施例6加速稳定性试验将实施例4制备的琥珀酸去甲文拉法辛晶型Ⅵ,以药用低密度聚乙烯袋封装后在40℃±2℃/75%±5%RH条件下,经加速6个月测试其稳定性,试验结果如下:表6.加速稳定性试验结果为了进一步证明本发明的晶型Ⅵ与CN100567253C公开的晶型Ⅰ、Ⅱ以及US20090234020公开方法制备的琥珀酸去甲文拉法辛一水合物晶型的区别,发明人制备了对比例1、2和3。并采用实施例7的方法制备琥珀酸去甲文拉法辛缓释片,然后比较它们在制成制剂后具有的不同性质(详见实施例7和8)。对比例1:琥珀酸去甲文拉法辛晶型Ⅰ的制备氮气环境下,将2090mL丙酮和660mL水加入到反应瓶中。搅拌并加入250g(0.95mol)去甲文拉法辛。搅拌30min。加入115g(0.98mol)琥珀酸。搅拌升温至60℃。搅拌30~60min。抽滤。滤液冷却至30~35℃,搅拌析晶4h。冷却至0‐5℃,搅拌析晶1h。抽滤。滤饼用0~5℃丙酮/水(24:76v/v,2×300mL)淋洗。30±5℃真空干燥12h。得产品325g,收率85.8%。对比例2:琥珀酸去甲文拉法辛晶型Ⅱ的制备反应瓶中加入去甲文拉法辛150g(1eq)、丙酮1.25L、琥珀酸39g(1eq)和水3.95L。搅拌升温至60℃,通过硅藻土垫过滤。滤垫用温丙酮194mL和水61.2mL的混合物洗涤。滤液 转移至洁净烧瓶中,100mL丙酮清洗。溶液温度28℃。冷却至23℃开始析晶。然后冰浴快速降温至0~5℃。搅拌析晶2h。抽滤。冷丙酮/水(24:76v/v,2×200mL)洗涤。滤饼35±5℃真空干燥48h。得白色固体162g,收率71.1%。对比例3:US20090234020公开方法制备琥珀酸去甲文拉法辛100g(1eq)去甲文拉法辛溶于异丙醇500mL。搅拌升温至回流。琥珀酸49.3g(1.1eq)。加入体系中,保温搅拌1h,5h内降温至25℃,搅拌过夜,抽滤,滤饼异丙醇淋洗2×50mL。112g,收率73.8%。实施例7:琥珀酸去甲文拉法辛缓释片的制备按理论收料1000片,称取实施例4、对比例1、对比例2和对比例3制得的琥珀酸去甲文拉法辛,以下步骤和处方制备缓释片。A:颗粒制备将微晶纤维素和羟丙甲纤维素过60目筛备用;称取琥珀酸去甲文拉法辛、微晶纤维素、羟丙甲纤维素和滑石粉,加入混合容器中,混合均匀,HSD100型料斗混合机,参数:转速10rpm,时间12min;加入内加的硬脂酸镁,混合均匀;HSD100型料斗混合机,参数:转速10rpm,时间4min;干法制粒,LGJ-80型干法制粒机,参数:筛网24目,送料转速20rpm,挤压转速8~12rpm,压力18~22MPa;总混:按处方比例将外加滑石粉和硬脂酸镁与部分颗粒预混后,加入羟丙甲纤维素,混合均匀;HSD100型料斗混合机,参数:转速10rpm时间6min。B:片芯制备颗粒含量测定,计算理论片重;以10mm圆形浅凹冲压片,片芯硬度6~12kg/cm2。C:薄膜包衣包衣液的配制:称取所需量纯化水,置入适宜容积的烧杯中,烧杯置于磁力搅拌器上,加入转子,搅拌;均匀的加入所需量欧巴代固体粉末;待所有的粉末全部加入后,降低搅拌速度,继续搅拌45min。包衣操作:测定恒流泵转速与流速的关系;设定包衣参数,主机转速调至片床可连续翻滚,但没有片芯被带过锅体最高点,开启预热;恒流泵转速使包衣液流速为10~15g/min、主机转速6~8rpm、温度控制45±5℃,进风转速1000~1400rpm,出风转速2000~2800rpm;待出风温度达到35~45℃后,开启蠕动泵开始喷浆,调节雾化压力0.1~0.2MPa;包衣液喷完后 干燥10~20min,停机。表7.琥珀酸去甲文拉法辛缓释片处方组成表8.各晶型制备缓释片收率晶型理论收料(片)实际收料(片)收率晶型Ⅰ100076376.3%晶型Ⅱ100081281.2%晶型Ⅵ100087587.5%US200902340201000‐‐US20090234020公开方法制备的琥珀酸去甲文拉法辛制片过程中黏冲现象十分严重,根本无法制成外表光洁的片剂,不适于工业生产。晶型Ⅵ制片收率优于晶型Ⅰ和晶型Ⅱ。实施例8:释放度考察缓控释制剂以其给药次数少、作用时间长、血药浓度平稳、生物利用度高等优点已成为制剂研究的一个基本领域。在缓控释制剂的研究过程中,往往需要对制剂释放度的相似性进行评价。文献报道缓控释制剂释放度相似性的评价方法很多,其中美国FDA推荐的一种评价试验制剂与对照制剂溶出度差异的方法。SFDA在2004年10月颁布的《化学药品补充申请技术指导原则(第一稿)》中亦提出采用f2法评价缓控释制剂释放度的相似性。目前上市的琥珀酸去甲文拉法辛缓释片是由辉瑞生产的,商品名为Pristiq的产品,由于制剂产品中存在大量辅料,因此目前无法获知琥珀酸去甲文拉法辛原料的晶型,作为仿制药企业,必然希望获得与原研产品更加一致的产品,因此,评价琥珀酸去甲文拉法辛缓释片释放度这一指标就尤为重要。本专利对不同晶型制备的琥珀酸去甲文拉法辛缓释片进行释放度考察。以含0.9%NaCl溶液900ml为释放介质,转速每分钟100转,于1h、2h、4h、6h、8h、10h、12h、16h、20h、24h、30h时间间隔取样10ml,滤过,取续滤液作为供试品溶液。另取琥珀酸去甲文拉法辛对照品适量,精密称定,用释放介质溶解并稀释制成每1ml中约含去甲文拉法辛55μg的溶液,取上述两种溶液,用高效液相测定法测定,并计算释放量。表9.各晶型缓释片累计释放度由释放度结果可知:晶型Ⅰ、晶型Ⅱ与市售制剂相比,相似因子分别为71.8、69.7;晶型Ⅵ制剂与市售制剂的相似因子为90.6,相似度更高。因此,可以得出结论:以相同的制备方法、相同工艺参数制备的片剂,由于晶型不同,制剂体外释放度的结果存在较大差异,晶型Ⅵ所获得的产品与市售制剂更加一致,与晶型Ⅰ、晶型Ⅱ相比更具有优势。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1