一系列新型氨基糖苷类化合物的制备与应用及其高产工程菌的构建方法与流程
本发明属于生物医药领域,具体涉及一系列新型氨基糖苷类化合物的制备与应用及其高产工程菌的构建方法。
背景技术:
伴随着细菌多重耐药性以及“超级细菌”的出现,理性设计具有新活性的氨基糖苷类抗生素俨然成为人们关注的焦点,而利用合成生物学和代谢工程的方法,是一种比化学合成和化学半合成更为有效、经济和环境友好的手段。前人对氨基糖苷类抗生素生物合成机制、分子水平的作用机制及耐药机制的全面深入揭示为我们利用合成生物学与代谢工程的思路对天然氨基糖苷类抗生素进行改造与创新提供了重要的基础和前提。
天然氨基糖苷类抗生素的结构中存在着两类重要及独特的天然结构单元:来自于丁酰苷菌素的AHBA侧链基团与来自于庆大霉素的多个修饰基团。前人在利用氨基糖苷类抗生素天然结构单元进行结构改造时,由于通过化学手段来利用庆大霉素修饰基团进行氨基糖苷类抗生素结构改造时需要克服更为复杂及困难的化学立体选择性问题,因而研究者们大多利用了AHBA侧链基团。
技术实现要素:
本发明的首要目的在于提供一系列基于庆大霉素(Gentamicin)与卡那霉素(Kanamycin)结构改造的新型氨基糖苷类化合物Genkamicins(Gentamicin与Kanamycin的组合)。
本发明的目的另一目的在于提供产生这些新型氨基糖苷类化合物的菌株。
本发明的目的再一目的在于提供产生这些新型氨基糖苷类化合物的制备方法。
本发明的目的还在于提供这些新型氨基糖苷类化合物的应用。
本发明的目的通过下述技术方案实现:
本发明提供的新型氨基糖苷类化合物包括:GK-A、GK-X2、GK-418、GK-JI-20B、GK-JI-20A、GK-C1、GK-C1a、GK-C2a、GK-C2、GK-C2b,其结构式如下:
一种产生上述新型氨基糖苷类化合物的工程菌株,为失活或缺失genM2基因且能异源表达kanE基因的庆大霉素产生菌。
一种高产GK-A的工程菌株,为同时失活或缺失genM2、genD1、genK基因且能异源表达kanE基因的庆大霉素产生菌。
一种高产GK-X2的工程菌株,为同时失活或缺失genM2、genQ、genK基因且能异源表达kanE基因的庆大霉素产生菌。
一种高产GK-418的工程菌株,为同时失活或缺失genM2、genQ基因且能异源表达kanE基因的庆大霉素产生菌。
一种高产GK-JI-20B的工程菌株,为同时失活或缺失genM2、genB3基因且能异源表达kanE基因的庆大霉素产生菌。
一种高产GK-C1a、GK-C2b和/或X2的工程菌株,为同时失活或缺失genM2、genK基因且能异源表达kanE基因的庆大霉素产生菌。
所述的庆大霉素产生菌优选为棘孢小单孢菌ATCC15835。
所述的kanE基因优选来源于卡那霉素链霉菌ATCC12853。
上述工程菌株的构建方法,包括如下步骤:将含有kanE基因的质粒导入相应基因失活或缺失的庆大霉素产生菌中构建得到;所述的含有kanE基因的质粒导入庆大霉素产生菌中能够表达kanE基因。
上述新型氨基糖苷类化合物的提取方法,包括如下步骤:将上述任一项工程菌株发酵培养,发酵液用硫酸酸化后离心,收集上清液;上清液通过阳离子交换树脂进行吸附,吸附后用氨水进行洗脱,洗脱液挥发氨气完全后再通过阴离子交换树脂吸附,收集洗脱液即得发酵液提取产物,发酵液提取产物中含有新型氨基糖苷类化合物。
上述新型氨基糖苷类化合物的纯化方法,包括如下步骤:
(1)液相色谱与蒸发光散射检测仪联用(HPLC-ELSD)从发酵液提取产物中纯化氨基 糖苷类化合物
将提取后的发酵产物溶于Milli-Q水中后过水系滤膜,过滤后取溶液进样,色谱条件为:A相为含0.2%(v/v)三氟乙酸的水溶液(即100mL溶液中含有三氟乙酸0.2mL),用氨水调节pH值至2.0;B相为乙腈;流速4mL/min,洗脱程序为2%-8%(体积百分数)B相梯度洗脱20min,柱后分流比为1:3,以1mL流速进入蒸发光散射检测器;检测器雾化池温度设置为105℃,漂移管温度设置为105℃,气体流速为2.8L/min。
(2)纯化后产物硫酸盐的制备
将HPLC-ELSD制备的三氟乙酸盐形式产物冻干,除去游离三氟乙酸后溶于Milli-Q水,每10mg产物以4mL阴离子树脂除去三氟乙酸根离子,再以浓硫酸将溶液pH调至5.5-5.8后冻干得硫酸盐形式氨基糖苷类化合物。
上述新型氨基糖苷类化合物在抗菌中的应用。
上述新型氨基糖苷类化合物在制备抗菌药物中的应用。
本发明的新型氨基糖苷类化合物具有良好的抗菌效果,GK-C1a和GK-C1的抗菌活性比卡那霉素有明显地提高,GK-C1a和GK-C1对于原本对卡那霉素不敏感的铜绿假单孢杆菌以及原本对卡那霉素及庆大霉素均不敏感的鲍曼不动杆菌都表现出很强的抗菌活性。本发明为抗生素耐药性问题提供了新的解决手段。
附图说明
图1是新型氨基糖苷类化合物Genkamicins生物合成示意图。
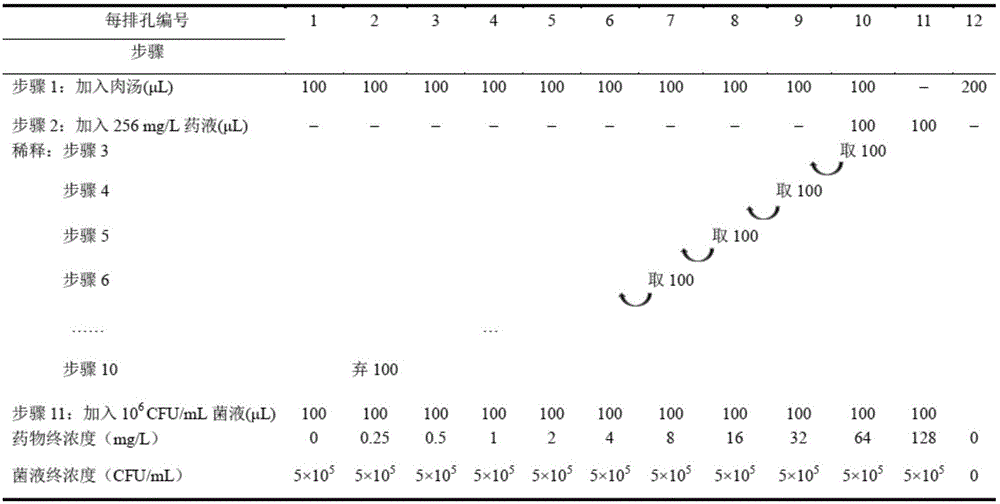
图2是微量肉汤稀释法药物抗菌敏感性试验方法的示意图。
图3是pWHU47质粒图谱。
图4是突变株ΔgenM2 PCR验证结果图,其中,泳道1-5分别是1Kb Ladder、pWHU47、ATCC15835、ΔgenM2、ΔgenM2。
图5是突变株ΔgenM2的LC-ESI-HRMS检测结果图,A:庆大霉素野生型(WT)产生菌ATCC15835,B:突变株ΔgenM2。
图6是pWHU155质粒图谱。
图7是突变株ΔgenM2::kanE的PCR验证结果图,其中,泳道1-3分别是1Kb Ladder、pWHU155、ΔgenM2::kanE。
图8是突变株ΔgenM2::kanE的LC-ESI-HRMS检测结果图,A:庆大霉素野生型产生菌ATCC15835,B:突变株ΔgenM2::kanE。
图9是突变株ΔgenS2ΔgenKΔgenM2 PCR验证结果图,其中,泳道1-5分别是1Kb Ladder、 pWHU47、ATCC15835、ΔgenS2ΔgenK、ΔgenS2ΔgenKΔgenM2。
图10是各突变株异源回补kanE的PCR验证结果图,其中,泳道1-5分别是1Kb Ladder、pWHU155、ΔgenS2ΔgenKΔgenM2::kanE、ΔgenQΔgenKΔgenM2::kanE、ΔgenD1ΔgenKΔgenM2::kanE。
图11是pWHU2649质粒图谱。
图12是突变株ΔgenD1ΔgenKΔgenM2 PCR验证结果图,其中,泳道1-5分别是1Kb Ladder、pWHU47、ATCC15835、ΔgenD1ΔgenKΔgenM2、ΔgenD1ΔgenK。
图13是突变株ΔgenQΔgenKΔgenM2 PCR验证结果图,其中,泳道1-5分别是1Kb Ladder、pWHU47、ATCC15835、ΔgenQΔgenK、ΔgenQΔgenKΔgenM2。
图14是突变株ΔgenM2ΔgenQ::kanE PCR验证结果图,其中,泳道1-5分别是1Kb Ladder、pWHU47、ATCC15835、ΔgenM2::kanE、ΔgenM2ΔgenQ::kanE。
图15是突变株ΔgenM2ΔgenB3::kanE PCR验证结果图,其中,泳道1-5分别是1Kb Ladder、pWHU47、ATCC15835、ΔgenM2::kanE、ΔgenM2ΔgenB3::kanE。
图16是突变株ΔgenM2ΔgenK::kanE PCR验证结果图,其中,泳道1-5分别是1Kb Ladder、pWHU47、ATCC15835、ΔgenM2::kanE、ΔgenM2ΔgenK::kanE。
图17是各突变株发酵产物的LC-ESI-HRMS检测结果图。
图18是化合物GK-C1的LC-ESI-HRMS结构鉴定结果图。
图19是化合物GK-C1a的LC-ESI-HRMS结构鉴定结果图。
图20是化合物GK-JI-20B的LC-ESI-HRMS结构鉴定结果图。
图21是化合物GK-418的LC-ESI-HRMS结构鉴定结果图。
图22是化合物GK-X2的LC-ESI-HRMS结构鉴定结果图。
具体实施方式
本发明以来源于卡那霉素生物合成中的糖基转移酶KanE置换庆大霉素生物合成中的同类功能元件GenM2,使庆大霉素产生菌合成出第三个糖环被置换的新三糖化合物中间体。而后,庆大霉素强大的后修饰系统对该中间体再进行不同的后续修饰,从而产生出一系列庆大霉素与卡那霉素不同结构单元进行组合后的新型氨基糖苷类化合物,并命名为Genkamicins(即Gentamicin与Kanamycin的组合)。这些新型氨基糖苷类化合物包括GK-A、GK-X2、GK-418、GK-JI-20B、GK-JI-20A、GK-C1、GK-C1a、GK-C2a、GK-C2、GK-C2b,其结构式及可能的生物合成途径如图1所示。
具体实施方式中所用实验材料及方法见下:
一、实验材料
1.菌株、质粒及引物如表1-3所示。
表1 实施例中所主要使用菌株
表2 实施例中所主要使用质粒
参考文献:
Huang et al.,2015:Huang C,Huang F,Eileen M,et al.2015.Delineating the biosynthesis of gentamicin X2,the common precursor of the gentamicin C antibiotic complex[J].Chem Biol,22(2):251-261.
Guo et al.,2014:Guo J,Huang F,Huang C,et al.2014.Specificity and promiscuity at the branch point in gentamicin biosynthesis[J].Chem Biol,21(5):608-618.
Sun et al.,2002:Sun Y,Zhou X,Liu J,Bao K,Zhang G,Tu G,Kieser T,Deng Z.2002.'Streptomyces nanchangensis',a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin,contains multiple polyketide gene clusters[J].Microbiology,148(2):361-371.
上述文献中详细记载了相应菌株或质粒的构建方法,本领域技术人员根据所记载的构建方法能够重复得到相应菌株或质粒。其中,pWHU77构建的具体过程为:
(1)以质粒pYH7为模板,扩增bla抗性基因片段和tsr抗性基因片段。其中,扩增bla抗性基因片段的引物为:
bla-F:ATGAGACAATAACCCTGATAAATGC,
bla-R:ACCGAGCTCCTTTTAAATTAAAAA;
扩增tsr抗性基因片段的引物为:
tsr-F:TAGGAGCTCACACCCCAGGAATC,
tsr-R:TTCGAGCTCTGATCATCACTGACGA。
(2)用PshAI酶切PIB139载体,回收4.9Kb的载体片段,将其与扩增的bla抗性基因片段连接构建得到重组质粒。再用SacI酶切该重组质粒和扩增得到的tsr抗性基因片段,连接SacI酶切后产物构建得到重组质粒pWHU77。
表3 实施例中所使用引物及其序列
2.培养基
(1)2×TY液体培养基:酵母提取物10g,胰蛋白胨16g,氯化钠5g,Milli-Q纯水1L,115℃高温灭菌30min。
(2)2×TY固体培养基:酵母提取物10g,胰蛋白胨16g,氯化钠5g,琼脂粉20g,Milli-Q纯水1L,115℃高温灭菌30min。
(3)ATCC172液体培养基:可溶性淀粉20g,酵母提取物5g,葡萄糖10g,酶解酪蛋白5g碳酸钙1g,Milli-Q纯水1L,115℃高温灭菌30min。
(4)ATCC172固体培养基:可溶性淀粉20g,酵母提取物5g,葡萄糖10g,酶解酪蛋白5g碳酸钙1g,琼脂粉20g,Milli-Q纯水1L,115℃高温灭菌30min。
(5)ABB13固体培养基:可溶性淀粉5g,大豆蛋白胨5g,碳酸钙3g,3-(N-吗啉)丙磺酸2.1g,盐酸硫胺0.01g,氧化亚铁0.012g,琼脂粉20g,Milli-Q纯水1L,氢氧化钾调节pH值至7.0,115℃高温灭菌30min。用前按1%的体积比加入过滤除菌后的1M氯化镁溶液。
(6)F50高产培养基:黄豆饼粉2.0%,可溶性淀粉3.0%,蛋白胨0.1%,葡萄糖0.3%,碳酸钙0.3%,硫酸铵0.03%,硝酸钾0.03%,氯化钴5ppm。
3.抗生素及其浓度如表4所示。
表4 抗生素及其使用浓度
二、实验方法
1.大肠杆菌与棘孢小单孢菌的培养
大肠杆菌的培养采用2×TY培养基,220rpm 37℃培养。棘孢小单孢菌的培养采用ATCC172培养基,200rpm 28℃培养。
2.大肠杆菌-棘孢小单孢菌属间结合转移
取在ATCC172固体培养基上生长3-4天的棘孢小单孢菌1平方厘米菌块于15mL ATCC172液体培养基中28℃220rpm培养2-3天至生长对数期作为接合转移受体菌。将在37℃220rpm过夜培养后以1:100(v/v)比例转接后培养2.5-3h至生长对数期的大肠杆菌ET12567/pUZ8002作为接合转移供体菌。将生长至对数期的供体菌与受体菌以分别5000 rpm×5min离心去除上清后收集菌体,并以2×TY液体培养基分别洗涤菌体2两次后,去除大部分上清,供体菌留存约1mL上清液,受体菌留存约5mL上清液,分别尽量吸取上部幼嫩菌体100μL后混合,再涂布于含有1%氯化镁的ABB13平板上,吹干后倒置于28℃恒温培养箱中培养12hr。再取1mL含常规浓度减半的筛选抗生素和萘啶酮酸的无菌水均匀地覆盖培养基表面,待培养基表面液体被吹干后将平板后封口,倒置于28℃恒温培养箱中培养。一般约120-144hr后可见明显接合转移子。
3.棘孢小单孢菌的发酵与发酵产物的提取与分离
(1)棘孢小单孢菌的发酵培养:
取在ATCC172固体培养基上培养3-4天的小单孢菌1cm2菌块接ATCC172液体培养基中,28℃200rpm培养3天,作为种子培养基。将种子培养基以1:20的比例转接至50mL发酵培养基中,28℃200rpm培养5-6天。
(2)棘孢小单孢菌发酵产物的提取与分离:
将发酵液以6M的浓硫酸调pH至2.0,震荡酸化4hr以上后,将酸化发酵液以6000rpm×10min、4℃离心,将上清液用滤纸过滤。将过滤后的上清液上样至2mL经乙腈及MillQ超纯水清洗过的DOWEX50-WX8-200阳离子交换树脂进行吸附。吸附后,以15mL 1N的氨水进行洗脱,收集氨水洗脱液,并于室温挥发氨气只至挥发完全。将挥氨后的洗脱液经4mL 717阴离子树脂除去色素等阴离子杂质,收集洗脱液即得发酵液提取产物。
4.氨基糖苷类化合物的纯化与制备
(1)HPLC-ELSD从发酵液提取产物中纯化氨基糖苷类化合物
将发酵液提取产物溶于Milli-Q超纯水中后过0.22μm水系滤膜,过滤后溶液取30-60μL进样,液相色谱仪为Dionex Ultimate3000,色谱柱采用Phenomenex公司的Synergi Hydro-RP80A半制备柱(250×10mm,4μm),色谱条件:A相为含0.2%三氟乙酸(TFA)的水溶液(用10%氨水调节pH值至2.0),B相为乙腈,流速4mL/min,洗脱程序为2%-8%B相梯度洗脱20min,柱后分流比为1:3,以1mL流速进入检测器。检测器为美国Alltech 2000ES,其雾化池温度设置为105℃,漂移管温度设置为105℃,气体流速为2.8L/min。
(2)纯化后产物硫酸盐的制备
将HPLC-ELSD制备的三氟乙酸盐形式产物冻干,尽量除去游离三氟乙酸后溶于Milli-Q超纯水,每10mg产物以4mL 717阴离子树脂除去三氟乙酸根离子,此时溶液应呈强碱性(pH>10),再以浓硫酸将溶液pH调至5.5-5.8后冻干即为硫酸盐形式。
5.氨基糖苷类化合物的LC-ESI-HRMS检测
将冻干后的发酵产物用1mL Milli-Q超纯水溶解后,以0.22μm水系微孔滤膜过滤后进样检测。检测仪器为Thermo公司出产的高分辨质谱仪LTQ-Obitrap XL。检测条件为:Phenomenex Luna C18(250×4.6mm,5μm)色谱柱,流动相为:A相为含0.2%三氟乙酸(TFA)的水溶液(用10%氨水调节pH值至2.0),B相为乙腈。洗脱程序为梯度洗脱:0-14min,98%A至94%A;14-16min,94%A至92%A;16-25min,92%A至85%A;25-26min,85%A至10%A;26-34min,10%A;34-35min,10%A至98%A;35-45min,98%A;流速为400μL/min。洗脱后的液体进过1:1分流后接入ESI-HRMS检测器,ESI选用正离子模式。经过优化后的离子源条件是:喷雾电压3.5kv,毛细管温度275℃,鞘气流速50L/hr,辅助气流速10L/hr,离子源温度为275℃,扫描范围m/z 300-600,碰撞能量35eV,样品进样量为10μL。
6.生物学活性——最小抑菌浓度值(Minimal inhibitory concentration,MIC)的测定
本方法参考美国临床和实验室标准协会(Clinical and Laboratory Standards Institute,CLSI)的需氧菌的稀释法抗菌药物敏感性试验,批准标准-第九版(Methods for Dilution Antimicrobial Susceptibility Test for Bacteria That Grow Aerobiacally,Approved Standard-Ninth Edition)中微量肉汤稀释法。
(1)抗菌药物原液的配置:
取常用测定浓度上限10倍数值为抗菌原液浓度,即1280mg/L。原液配置好后用0.22μm微孔滤膜过滤除菌后,小量分装备用,剩余原液-20℃保存。
(2)测试菌的接种:
培养基的配置:菌株活化培养基选用哥伦比亚血平板。抑菌试验选取MH肉汤,测定对铜绿假单胞菌的MIC值时补充二价阳离子(镁离子25mg/L和钙离子50mg/L)。
测试菌液的准备:将测试菌以平板划线法在哥伦比亚血平板上进行活化,37℃培养观察16-18hr待单克隆菌株生长后,挑取几个单一菌落接直接悬浮与MH培养基中至0.5麦氏比浊标准,再用肉汤以1:100稀释(含菌量约106CFU/mL)备用。比浊标准:0.048mol/L氯化钡0.5mL加0.18mol/L硫酸溶液99.5mL,混匀后用530nm波长调整期浊度,使吸收值为0.1,该浊度为0.5麦氏比浊标准,相当于5×(107-108CFU/mL)。
抗菌药物的稀释:将抗菌药物用MH肉汤以二倍稀释法将第2至第11个孔稀释成下列浓度:256mg/L、128mg/L、64mg/L、32mg/L、16mg/L、8mg/L、4mg/L、2mg/L、1mg/L、0.5mg/L,稀释方法见图2。
测试菌的接种:取稀释好的测试菌液依次由抗菌药物低浓度至高浓度从第1至第11个孔中每孔接种100μL,则每孔最终接种菌量约为5×105CFU/mL,每排药物最终浓度为128mg/L、 64mg/L、32mg/L、16mg/L、8mg/L、4mg/L、2mg/L、1mg/L、0.5mg/L、0.25mg/L、0.125mg/L、0mg/L物与菌液混匀后,加盖并用parafilm封口膜密封后置于37℃培养观察18-20hr,操作步骤如见图2。
(3)结果判断:
在黑色背景光源下观察肉汤出现浑浊,孔底出现沉淀为标准判断测试菌是否生长,无测试菌生长的所含最低抗菌药物浓度即为MIC值。为使结果更快速清晰,可在每孔中加入5g/L氯化三苯四氮唑(TTC)5μL,35℃孵育1-3hr后呈红色的为有测试菌生长。
下面结合实施例对本发明做进一步详细的描述,但本发明的实施方式不限于此。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
实施例1产生新型氨基糖苷类化合物工程菌株的构建
产生新型氨基糖苷类化合物工程菌株为失活或缺失genM2基因且能异源表达kanE基因的庆大霉素产生菌。
(1)genM2同框敲除重组质粒pWHU47的构建
1)以庆大霉素野生型产生菌棘孢小单孢菌(Miocromonospora echinospora)ATCC15835的染色体DNA为模板,以genM2-L-F和genM2-L-R为引物,在genM2基因上游扩增出2344bp片段作为genM2同框敲除同源左臂genM2-L。以genM2-R-F和genM2-R-R为引物,在genM2基因下游扩增出2183bp片段作为genM2同框敲除同源右臂genM2-R。
2)将同源左臂genM2-L利用扩增引物中所引入的酶切位点NdeI与EcoRI克隆至pUC18,得到重组质粒pUC18-genM2-L,并经酶切和测序验证正确。同样地,将同源右臂genM2-R利用扩增引物中所引入的酶切位点EcoRI与HindIII克隆至pUC18,得到重组质粒pUC18-genM2-R,经酶切和测序验证正确。
3)将pUC18-genM2-L和pUC18-genM2-R分别用NdeI与EcoRI、EcoRI与HindIII经三片段连接方式同时克隆至pYH7,经酶切验证正确后得到genM2同框敲除重组质粒并命名为pWHU47(图3)。
(2)genM2同框敲除菌株ΔgenM2的构建
将genM2同框敲除重组质粒pWHU47转入大肠杆菌ET12567/pUZ8002,以阿泊拉霉素、卡那霉素及氯霉素作为抗性筛选标记进行筛选,将筛选到的经质粒提取及酶切验证正确的转化子ET12567/pUZ8002+pWHU47作为接合转移供体菌,以庆大霉素野生型产生菌ATCC15835作为接合转移受体菌,按照上述结合转移方法将pWHU47导入其中。再利用阿泊拉霉素为筛选标记,筛选到具有阿泊拉霉素抗性的接合转移子后将其进行松弛培养(松弛 培养:在含有1%MgCl2的培养平板上划单克隆培养),将松弛培养获得的单克隆经在含有阿泊拉霉素和非抗平板上进行影印培养,筛选到对阿泊拉霉素敏感的菌株,这些菌株为可能的双交换突变株或野生型菌株。利用验证引物genM2-CK-F和genM2-CK-R,以其基因组DNA为模板进行PCR筛选和验证,理论阳性扩增条带为639bp。经PCR筛选和验证正确(图4)的菌株为双交换突变株即genM2同框敲除菌株ΔgenM2,进一步通过Southern blot(Southern印迹杂交)证实突变株ΔgenM2构建成功。
将突变株ΔgenM2同对照组庆大霉素野生型产生菌ATCC15835进行发酵,并对其发酵产物进行LC-ESI-HRMS分析以测试ΔgenM2是否终止了庆大霉素的产生,结果如图5所示。结果显示突变株ΔgenM2终止了包括了庆大霉素所有三糖中间产物GentamicinA2、GentamicinA、GentamicinX2、G418、Gentamicin JI-20B、Gentamicin JI-20A及Gentamicin C1、Gentamicin C1a、Gentamicin C2、Gentamicin C2a、Gentamicin C2b五种终产物的产生,同时大量积累了二糖中间体巴龙霉胺。
(3)产生新型氨基糖苷类化合物工程菌株的构建
将基因kanE异源回补ΔgenM2突变株,其构建过程如下:
1)kanE异源表达质粒pWHU77-kanE的构建
以卡那霉素产生菌卡那霉素链霉菌(Streptomyces kanamyceticus)ATCC12853的基因组DNA为模板,以kanE-F和kanE-R为引物,PCR扩增出包含完整kanE在内的1259bp片段,并通过扩增引物中所引入的酶切位点NdeI/EcoRI将其克隆至pWHU77,获得重组质粒pWHU77-kanE,经酶切和测序验证正确后命名为pWHU155(图6)。
2)基因kanE异源回补ΔgenM2突变株ΔgenM2::kanE的构建
将pWHU155转入大肠杆菌ET12567/pUZ8002,以氨苄青霉素、卡那霉素及氯霉素作为抗性筛选标记进行筛选,将筛选到的经质粒提取及酶切验证正确的转化子ET12567/pUZ8002+pWHU155作为接合转移供体菌,以突变株ΔgenM2作为接合转移受体菌,通过结合转移方法将pWHU155导入其中。再利用硫链丝菌素作为筛选标记,筛选到具有硫链丝菌素抗性的接合转移子即为可能的kanE异源表达突变株ΔgenM2::kanE。利用验证引物pWHU77-CK-F和pWHU77-CK-R为引物,以其基因组DNA为模板,对其进行PCR验证,理论上,突变株ΔgenM2::kanE应扩增得到1651bp条带。PCR验证(图7)证实得到突变株ΔgenM2::kanE,并将其命名为ΔgenM2::pWHU155。
将突变株ΔgenM2::pWHU155与野生型ATCC15835及ΔgenM2对照组进行发酵,并对其发酵产物进行LC-ESI-HRMS分析,对图1中的Genkamicins系列产物的分子离子峰进行抽提, 并对抽提到的分子离子进行二级质谱检测,结果见图8。结果显示突变株ΔgenM2::pWHU155产生了Genkamicins系列产物:GK-A2、GK-A、GK-X2、GK-JI20A、GK-JI20B、GK-C1a、GK-C2、GK-C2a、GK-C2b;而对照组ΔgenM2及ATCC15835发酵产物中均不能被检测到这些新化合物。对比分析突变株ΔgenM2::pWHU155中Genkamicins系列产物的产量与庆大霉素野生型产生菌中庆大霉素对应结构的产量发现:突变株中Genkamicins系列产物之间的比例与野生型菌株中庆大霉素各产物之间比例相近,但突变株中Genkamicins系列产物产量较野生型菌株中对应结构庆大霉素产量均有少量下降。
实施例2高产3’-脱氨基-3’-羟基-卡那霉素C(命名为GK-A2)工程菌株的构建
高产GK-A2工程菌株为同时失活或缺失genM2、genS2、genK基因且能异源表达kanE基因的庆大霉素产生菌。
(1)ΔgenS2ΔgenKΔgenM2突变株的构建
以实施例1构建好的ET12567/pUZ8002+pWHU47作为接合转移供体菌,以突变株ΔgenS2ΔgenK为出发菌株与接合转移受体菌,通过结合转移方法将pWHU47导入其中,筛选到可能为双交换突变株或野生型菌株的菌株,进一步通过PCR验证(图9)筛选到双交换突变株ΔgenS2ΔgenKΔgenM2,具体过程同实施例1ΔgenM2的构建,进一步通过Southern blot证实突变株ΔgenS2ΔgenKΔgenM2构建成功。
再将实施例1构建好的ET12567/pUZ8002+pWHU155作为接合转移供体菌,以突变株ΔgenS2ΔgenKΔgenM2为出发菌株与接合转移受体菌,通过结合转移方法将pWHU155导入其中。突变株ΔgenS2ΔgenKΔgenM2::kanE的具体筛选方法与PCR验证方法同实施例1ΔgenM2::kanE的构建,PCR验证结果见图10,筛选到的突变株ΔgenS2ΔgenKΔgenM2::kanE命名为ΔgenS2ΔgenKΔgenM2::pWHU155。
实施例3高产Genkamicn A(GK-A)工程菌株的构建
高产GK-A工程菌株为同时失活或缺失genM2、genD1、genK基因且能异源表达kanE基因的庆大霉素产生菌。
(1)genM2 inΔgenD1ΔgenK同框敲除重组质粒pWHU2649的构建
1)以突变株ΔgenD1ΔgenK的染色体DNA为模板,在genM2基因上游以genM2 inΔgenD1-L-F和genM2 inΔgenD1-L-R为引物扩增出2355bp片段为同框敲除同源左臂genM2 inΔgenD1-L。在genM2基因下游以genM2 inΔgenD1-R-F和genM2 inΔgenD1-R-R为引物扩增出2198bp片段为同框敲除同源右臂genM2 inΔgenD1-R。
其中,突变株ΔgenD1ΔgenK的构建方法如下:
将genK同框敲除重组质粒pWHU1转入大肠杆菌ET12567/pUZ8002,以阿泊拉霉素、卡那霉素及氯霉素作为抗性筛选标记进行筛选,将筛选到的经质粒提取及酶切验证正确的转化子ET12567/pUZ8002+pWHU1作为接合转移供体菌,以突变株ΔgenD1作为接合转移受体菌,按照上述结合转移方法将pWHU1导入其中。再利用阿泊拉霉素为筛选标记,筛选到具有阿泊拉霉素抗性的接合转移子后将其在含有1%MgCl2的培养板上划单克隆培养(松弛培养),将获得的单克隆经在含有阿泊拉霉素和非抗平板上进行影印培养,筛选到对阿泊拉霉素敏感的菌株,这些菌株为可能的双交换突变株或野生型菌株。利用验证引物genK-CK-F和genK-CK-R,以其基因组DNA为模板进行PCR筛选和验证,理论阳性扩增条带为585bp。经PCR筛选和验证正确的菌株为双交换突变株,即genD1与genK的同框敲除菌株ΔgenD1ΔgenK。
2)同源左右臂的克隆及三片段连接方法与实施例1中pWHU47的构建方法相同,经酶切和测序验证正确后得到genM2 inΔgenD1ΔgenK同框敲除重组质粒并命名为pWHU2649(图11)。
(2)ΔgenD1ΔgenKΔgenM2突变株的构建
将pWHU2649转入大肠杆菌ET12567/pUZ8002中,得到转化子ET12567/pUZ8002+pWHU2649作为接合转移供体菌,以突变株ΔgenD1ΔgenK为出发菌株与接合转移受体菌,通过结合转移方法将pWHU2649导入其中,筛选到可能为双交换突变株或野生型菌株的菌株,具体方法同实施例1ΔgenM2的构建。进一步以genM2 inΔgenD1-CK-R和genM2 inΔgenD1-CK-F为验证引物,对其进行PCR验证,理论阳性扩增条带为510bp。经PCR筛选和验证正确(图12)的菌株为双交换突变株ΔgenD1ΔgenKΔgenM2,进一步通过Southern blot证实突变株ΔgenD1ΔgenKΔgenM2构建成功。
(3)高产GK-A工程菌株的构建
将基因kanE异源回补ΔgenD1ΔgenKΔgenM2突变株,其构建过程如下:
以实施例1中构建好的ET12567/pUZ8002+pWHU155作为接合转移供体菌,以突变株ΔgenD1ΔgenKΔgenM2为出发菌株与接合转移受体菌,通过结合转移方法将pWHU155导入其中。突变株ΔgenD1ΔgenKΔgenM2::kanE的具体筛选方法与PCR验证方法同实施例1ΔgenM2::kanE的构建,将PCR验证正确(图10)的突变株ΔgenD1ΔgenKΔgenM2::kanE命名为ΔgenD1ΔgenKΔgenM2::pWHU155。
实施例4高产Genkamicn X2(GK-X2)工程菌株的构建
高产GK-X2工程菌株为同时失活或缺失genM2、genQ、genK基因且能异源表达kanE 基因的庆大霉素产生菌。
以实施例1构建好的ET12567/pUZ8002+pWHU47作为接合转移供体菌,以突变株ΔgenQΔgenK为出发菌株与接合转移受体菌,通过结合转移方法将pWHU47导入其中,筛选到可能为双交换突变株或野生型菌株的菌株,进一步通过PCR验证(图13)筛选到双交换突变株ΔgenQΔgenKΔgenM2,进一步通过Southern blot证实突变株ΔgenQΔgenKΔgenM2构建成功,具体过程同实施例1ΔgenM2的构建。
再将实施例1构建好的ET12567/pUZ8002+pWHU155作为接合转移供体菌,以突变株ΔgenQΔgenKΔgenM2为出发菌株与接合转移受体菌,通过结合转移方法将pWHU155导入其中。突变株ΔgenQΔgenKΔgenM2::kanE的具体筛选方法与PCR验证方法同实施例1ΔgenM2::kanE的构建,将PCR验证正确(图10)的突变株ΔgenQΔgenKΔgenM2::kanE命名为ΔgenQΔgenKΔgenM2::pWHU155。
实施例5高产Genkamicn 418(GK-418)工程菌株的构建
高产GK-418工程菌株为同时失活或缺失genM2、genQ且能异源表达kanE基因的庆大霉素产生菌。可以在突变株ΔgenQ的基础上同框敲除genM2基因再进一步异源回补kanE基因,也可以直接在实施例1构建的突变株ΔgenM2::kanE的基础上同框敲除genQ基因得到高产GK-418工程菌株,本实施例工程菌株的构建采用了第二个策略。
以ET12567/pUZ8002+pYH286(转入pYH286质粒的大肠杆菌ET12567/pUZ8002)作为接合转移供体菌,以突变株ΔgenM2::kanE为出发菌株与接合转移受体菌,通过结合转移方法将pYH286导入其中,按照实施例1中的方法筛选双交换突变株。筛选到可能的双交换突变株后,利用验证引物genQ-CK-F和genQ-CK-R,以其基因组DNA为模板进行PCR筛选和验证,理论阳性扩增条带为522bp。经PCR筛选和验证(图14)正确的菌株为双交换突变株ΔgenM2ΔgenQ::kanE,进一步通过Southern blot证实突变株ΔgenM2ΔgenQ::kanE构建成功,将其命名为ΔgenM2ΔgenQ::pWHU155,。
实施例6高产Genkamicn JI-20B(GK-JI-20B)工程菌株的构建
高产GK-JI-20B工程菌株为同时失活或缺失genM2、genB3且能异源表达kanE基因的庆大霉素产生菌。可以在突变株ΔgenB3的基础上同框敲除genM2基因再进一步异源回补kanE基因,也可以直接在实施例1构建的突变株ΔgenM2::kanE的基础上同框敲除genB3基因得到高产GK-JI-20B工程菌株,本实施例工程菌株的构建采用了第二个策略。
以ET12567/pUZ8002+pWHU5(转入pWHU5质粒的大肠杆菌ET12567/pUZ8002)作为接合转移供体菌,以突变株ΔgenM2::kanE为出发菌株与接合转移受体菌,通过结合转移方法 将pWHU005导入其中,按照实施例1中的方法筛选双交换突变株。筛选到可能的双交换突变株后,利用验证引物genB3-CK-F和genB3-CK-R,以其基因组DNA为模板进行PCR筛选和验证,理论阳性扩增条带为587bp。经PCR筛选和验证(图15)正确的菌株为双交换突变株ΔgenM2ΔgenB3::kanE,进一步通过Southern blot证实突变株ΔgenM2ΔgenB3::kanE构建成功,将其命名为ΔgenM2ΔgenB3::pWHU155。
实施例7高产Genkamicn C1a(GK-C1a)和Genkamicn C2b(GK-C2b)工程菌株的构建
高产GK-C1a和GK-C2b工程菌株为同时失活或缺失genM2、genK且能异源表达kanE基因的庆大霉素产生菌。可以在突变株ΔgenK的基础上同框敲除genM2基因再进一步异源回补kanE基因,也可以直接在实施例1构建的突变株ΔgenM2::kanE的基础上同框敲除genK基因得到高产GK-C1a和GK-C2b工程菌株,本实施例工程菌株的构建采用了第二个策略。
以ET12567/pUZ8002+pWHU1(转入pWHU1质粒的大肠杆菌ET12567/pUZ8002)作为接合转移供体菌,以突变株ΔgenM2::kanE为出发菌株与接合转移受体菌,通过结合转移方法将pWHU1导入其中,按照实施例1中的方法筛选双交换突变株。筛选到可能的双交换突变株后,利用验证引物genK-CK-F和genK-CK-R,以其基因组DNA为模板进行PCR筛选和验证,理论阳性扩增条带为625bp。经PCR筛选和验证(图16)正确的菌株为双交换突变株ΔgenM2ΔgenK::kanE,进一步通过Southern blot证实突变株ΔgenM2ΔgenK::kanE构建成功,将其命名为ΔgenM2ΔgenK::pWHU155。
实施例8菌株发酵产物的LC-ESI-HRMS分析
将上述构建的各高产菌株及对照组ΔgenM2::kanE一起进行50mL小规模平行发酵与产物提取,并对其发酵产物进行LC-ESI-HRMS检测,对各突变株中Genkamicins系列产物的分子离子峰进行了抽提,确认了各组分二级碎片结构,结果如图17所示。
(1)突变株ΔgenS2ΔgenKΔgenM2::kanE按预期积累了GK-A2,其产量相较于ΔgenM2::kanE提高了约10倍,且无后续干扰三糖产物的产生,如图17中(A)和(B)。
(2)突变株ΔgenQΔgenKΔgenM2::kanE按预期积累了GK-X2,但其产量相较于ΔgenM2::kanE只提高了约3倍,如图17中(A)和(C)。
(3)突变株ΔgenD1ΔgenKΔgenM2::kanE按预期积累了GK-A,其产量相较于ΔgenM2::kanE提高了约10倍,如图17中(A)和(D),其下游的干扰产物已不再产生。
(4)突变株ΔgenM2ΔgenQ::kanE按预期积累了GK-418,且其产量相较于ΔgenM2::kanE提高了约20倍,其主要干扰产物GK-X2及上游产物GK-A2和GK-A的产量基本没有提高, 如图17中(A)和(E)。
(5)突变株ΔgenM2ΔgenB3::kanE按预期积累了GK-JI-20B,且其产量相较于ΔgenM2::kanE提高了约70倍,其主要干扰产物GK-418及上游产物GK-A2与GK-A的产量只较有小幅度的上升,如图17中(A)和(F);
(6)突变株ΔgenM2ΔgenK::kanE按预期积累了GK-C1a与GK-C2b,且它们的产量相较于ΔgenM2::kanE分别提高了约35倍和3倍,基本没有干扰产物;同时也发现其上游产物GK-X2产量有近100倍的大幅提高,如图17中(A)和(G),该突变株可用于GK-C1与GK-C2b的纯化制备,也可以用于GK-X2的制备。
实施例9 GK-C1、GK-C1a、GK-X2、GK-JI-20B、GK-418的制备与生物学活性测定
(1)突变株的大量发酵与产物粗提取
将ΔgenM2::kanE、ΔgenM2ΔgenK::kanE、ΔgenM2ΔgenB3::kanE、ΔgenM2ΔgenQ::kanE、ΔgenQΔgenKΔgenM2::kanE等突变株按如下方法进行大量发酵与产物粗提取:
将在50mL×4ATCC172培养基中28℃培养4天的种子发酵培养基以1:10(v/v)的比例转接至250mL×8的F50高产培养基中28℃发酵5天。将发酵完成的发酵液以6M浓硫酸酸化至pH 2.0,并持续搅拌酸化6hr。将酸化破菌后的发酵液以5000rpm×30min离心过滤取上清液,并加入70mL DOWEX50-WX8-200阳离子交换树脂过夜吸附。将吸附后的阳离子交换树脂装柱,并首先以约5倍柱体积的0.3M盐酸0.05M氯化铵溶液(含盐酸0.3M、氯化铵0.05M的溶液)冲洗树脂以出去未吸附或吸附不强的杂质,随后以5倍柱体积的Milli-Q超纯水冲洗除去残留在柱内的上述溶液。再以7-8倍柱体积即500mL 1M的氨水对吸附柱进行洗脱,并将洗脱液进行过夜挥氨处理。将挥氨处理后的洗脱液上样至装载有30mL 717阴离子交换树脂的吸附柱,以除去大部分色素等阴离子杂质,收集洗脱液,并将洗脱液浓缩至15mL得到发酵提取浓缩液。
通过HPLC-ELSD从发酵液发酵提取浓缩液中制备各目标产物,将所收集流份冻干称重,从突变株ΔgenM2::kanE 2mL发酵提取浓缩液中制得GK-C1三氟乙酸盐形式产物17.3mg;从突变株ΔgenM2ΔgenK::kanE 2.2mL发酵提取浓缩液中制得GK-C1a与GK-X2三氟乙酸盐形式产物分别为14.3mg与13.9mg;从突变株ΔgenM2ΔgenB3::kanE 1.3mL发酵提取浓缩液中制得GK-JI-20B三氟乙酸盐形式产物18.7mg;从突变株ΔgenM2ΔgenQ::kanE 1.7mL发酵提取浓缩液中制得GK-418三氟乙酸盐形式产物19.3mg;从突变株ΔgenQΔgenKΔgenM2::kanE 1.2mL发酵提取浓缩液中制得GK-X2三氟乙酸盐形式产物8.9mg。
将这些产物进行了LC-ESI-HRMS及1H、13C、1H-1H COSY、HSQC、HMBC、NOESY 的NMR检测,经结构鉴定,上述各组分产物结构均图1中结构相符,其LC-ESI-HRMS如图18-22所示,1H、13C NMR检测波谱数据表5-9。
表5 化合物GK-C1的1H和13C NMR波谱数据(600M,D2O,ppm)
表6 化合物GK-C1a的1H和13C NMR波谱数据(600M,D2O,ppm)
表7 化合物GK-JI-20B的1H和13C NMR波谱数据(600M,D2O,ppm)
表8 化合物GK-418的1H和13C NMR波谱数据(600M,D2O,ppm)
表9 化合物GK-X2的1H和13C NMR波谱数据(600M,D2O,ppm)
(2)生物活性测定
选取临床最常见的五种致病菌标准菌株(大肠埃希氏杆菌ATCC25922、金黄色葡萄球菌ATCC29213、铜绿假单孢杆菌ATCC27853、鲍曼不动杆菌ATCC19606、肺炎克雷伯氏菌ATCC70603)作为测试对象,对GK-C1、GK-C1a、GK-X2、GK-JI-20B、GK-418,连同作为对照的卡那霉素、庆大霉素及庆大霉素各单组分(C1a、C1、G418、X2)进行最小抑菌浓度(MIC)测试,测试结果如表10。
表10 微量肉汤稀释法MIC值测试结果
注:Gens:庆大霉素混合物,Kans:卡那霉素混合物。
GK-C1与GK-C1a对上述所有测试菌均表现出较卡那霉素更好的活性,GK-JI-20B、GK-418、GK-X2中,除GK-JI-20B对于大肠埃希氏菌及金黄色葡萄球菌活性均高于卡那霉素外,其它活性均与卡那霉素持平或略低于卡那霉素。GK-C1、GK-C1a、GK-JI-20B、GK-418、GK-X2对上述测试菌活性绝大多数弱于庆大霉素混合物。GK-C1与GK-C1a获得了对于卡那霉素不敏感的铜绿假单孢杆菌及对卡那霉素与庆大霉素均不敏感的鲍曼不动杆菌很强的活性——GK-C1a对铜绿假单孢杆菌MIC值为0.25-0.5μg/mL,对鲍曼不动杆菌MIC值为1-2μg/mL,GK-C1对鲍曼不动杆菌的MIC值为2-4μg/mL。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!