氟烷基芴衍生物的制作方法
本发明涉及具有使之适用于生产电气器件(electrical device)的电子和光电性质的新型化合物。本发明进一步涉及包括含这些化合物的层的电子器件(electronic device),其中这些化合物充当电荷传输材料或光致发光材料。
背景技术:
有机发光二极管(OLED)是其中发光电致发光材料是响应电流发光的有机材料的膜的发光二极管。OLED的发光有机层夹于两层电接触层之间。为了提高的效率,除了发光层之外,OLED器件可以在发光层和电接触层之间包括电荷传输材料层。这些电荷传输层可以包含空穴或电子传输材料。这些电荷传输材料能够允许载荷空穴和电子迁移通过发光层,由此有助于它们的组合以形成称为激发子的束缚态。激发子中的电子在适当的时候通过发射对于OLED器件是可见光区域中最常见频率的辐射而松弛转入低能态。
对开发具有适合用于制作OLED器件的改进性能的新材料存在极大的兴趣。例如,充当发射体、电子传输器和空穴传输器的材料,特别令人令人感兴趣。多年来已经开发了许多材料以试图产生改进的OLED器件,特别是具有最佳光输出、能源效率和寿命的器件。此外,另外显著的目标是实现允许器件制作方法简化的材料。尽管存在现有的材料,但对于具有如上述确定的具有性能的优异组合而适用于OLED器件及其它电子器件制作的性能的材料仍存需要。
已知可以适用于制作有机电子器件的是以下通式的一些反应性液晶元(能够化学交联至聚合物基质的液晶材料):
B-S-A-S-B
其中,A表示含有在C-9上用两个烷基基团取代的芴的直链芳族分子核芯,S表示柔性间隔单元而B表示交联基团如甲基丙烯酸酯基团。如果B表示光可交联的基团时,这尤其是这种情况,因为随后该材料实质上充当了光刻胶的作用,也就是说,这些材料的薄层可以通过图案化暴露于光,尤其是UV光而图案化成有用的电子结构。
此外,如果直链芳族核芯A是发光性的,则这些反应性液晶元材料可以图案化至电致发光器件如有机发光二极管(OLED)和有机二极管激光器的活性发光层中。然而,B-S-A-S-B结构的工作OLED器件都表现出低得令人失望的寿命。
本发明的目的在于提供用于电子器件的新型含芴材料,该材料克服或显著降低与现有芴衍生物相关的问题。
技术实现要素:
在本发明的第一方面中,提供了式(I)的化合物
D-S1-A-S2-B1, 式(I)
其中:
A表示-Ar1-(FL-Ar2)n-并包括1至8个FL基团;
Ar1和Ar2在每次出现时独立地选自包括Ara和键的组中;
Ara表示包括1个芳族、杂芳族或FL部分,或者2、3、4或5个通过单键互连的芳族、杂芳族和/或FL部分的双自由基(diradical);
n是1至8的整数;
FL是在C-2和C-7处通过共价键引入链中的以下结构的芴部分
每个FL部分的R基团是相同的并选自由直链或支链的非手性C1-C14烷基、C1-C14卤代烷基、C1-C14氟烷基、C2-C14烯基基团组成的组中,可选地,其中1、2、3、4或5个CH2基团被氧代替,条件是在R基团中不存在缩醛、缩酮、过氧化物或乙烯基醚;
D表示可交联基团或,当B1表示氢时,D表示-B2-S3-B3-S1a-A-S2a-B1a、-S3(B2)-B3-S1a-A-S2a-B1a、-S3(B2)(B3)-S1a-A-S2a-B1a、-S3(B2)(B3)或其中链左端处的破折号表示至S1的连接点的可交联基团;
B1表示可交联基团或氢原子;
B1a表示可交联基团或氢原子;
B2和B3各自表示可交联基团;
S1、S2、S1a和S2a是柔性连接基团;和
S3是间隔基团。
在一个实施方式中,提供了式Ia的化合物,其中每个FL部分的R基团是相同的并选自由直链或支链的非手性C2-C10烷基、C2-C10卤代烷基、C2-C10氟烷基、C2-C10烯基基团组成的组中,可选地,其中1、2或3个CH2基团被氧代替,在R基团中不存在缩醛、缩酮、过氧化物或乙烯基醚,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、Ar1、Ar2、FL和n如上对于式(I)的化合物所定义的。
在一个实施方式中,提供了式Ib的化合物,其中每个FL部分的R基团是相同的并选自由直链C2-C10烷基、C2-C10卤代烷基、C2-C10氟烷基、C2-C10烯基基团组成的组中,可选地,其中1、2或3个CH2基团被氧代替,在R基团中不存在缩醛、缩酮、过氧化物或乙烯基醚,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、Ar1、Ar2、FL和n如上对于式(I)的化合物所定义的。
在一个实施方式中,提供了式Ic的化合物,其中每个FL部分的R基团是相同的并选自由直链或支链的非手性C2-C10烷基和C2-C10氟烷基组成的组中,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、Ar1、Ar2、FL和n如上对于式(I)的化合物所定义的。
在一个实施方式中,提供了式Id的化合物,其中每个FL部分的R基团是相同的并选自由直链或支链的非手性C1-C14烷基、C2-C10烷基、C3-C8烷基、甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、癸基组成的组中,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、Ar1、Ar2、FL和n如上对于式(I)的化合物所定义的。
在一个实施方式中,提供了式Ie的化合物,其中每个FL部分的R基团是相同的并选自由直链C1-C14烷基、C2-C10烷基、C3-C8烷基、甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、癸基组成的组中,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、Ar1、Ar2、FL和n如上对于式(I)的化合物所定义的。
在一个实施方式中,提供了式If的化合物,其中n是1至6,可选地3至6的整数,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、Ar1、Ar2、FL和R如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)或(Ie)的化合物所定义的。
在一个实施方式中,提供了式Ig的化合物,其中n是3至6的整数,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、Ar1、Ar2、FL和R如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)或(Ie)的化合物所定义的。
在一个实施方式中,提供了式Ih的化合物,其中Ar1和Ar2在每次出现时独立地选自包括Ara和键的组中,其中Ara表示包括1个芳族、杂芳族或FL部分,或者2或3个通过单键互连的芳族、杂芳族和/或FL部分的双自由基,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)或(Ig)的化合物所定义的。
在一个实施方式中,提供了式Ii的化合物,其中Ar1和Ar2在每次出现时独立地选自包括Ara和键的组中,其中Ara表示包括1个芳族、杂芳族或FL部分,或者2或3个通过单键互连的芳族、杂芳族和/或FL部分的双自由基,并且其中芳族的和杂芳族的部分在每次出现时独立地选自由以下组成的组中:1,4-亚苯基、联苯-4,4’-二基、三联苯-4,4"-二基、萘-1,4-二基、萘-2,6-二基、噻吩-2,5-二基、嘧啶-2,5-二基、吡啶-2,5-二基、苝-3,10-二基、芘-2,7-二基、2,2’-二噻吩-5,5’-二基、噁唑-2,5-二基、1,3,4-噁二唑-2,5-二基、噻吩并[3,2-b]噻吩-2,5-二基、二噻吩并[3,2-b:2’,3’-d]噻吩-2,6-二基、二苯并噻吩-3,7-二基、苯并[1,2-b:4,5-b’]二[1]苯并噻吩-3,9-二基、噻唑并[5,4-d]噻唑-2,5-二基、噁唑并[5,4-d]噁唑-2,5-二基、噻唑并[5,4-d]噁唑-2,5-二基、噻唑并[4,5-d]噻唑-2,5-二基、噁唑并[4,5-d]噁唑-2,5-二基、噻唑并[4,5-d]噁唑-2,5-二基、2,1,3-苯并噻二唑-4,7-二基、4-噻吩-2-基-2,1,3-苯并噻唑-7,5’-二基、4,7-二噻吩-2-基-2,1,3-苯并噻唑-5’,5”-二基、咪唑并[4,5-d]咪唑-2,5-二基、4-烷基-1,2,4-三唑-3,5-二基、4-芳基-1,2,4-三唑-3,5-二基、4-苯基-1,2,4-三唑-3,5-二基、4-(对叔丁基苯基)-1,2,4-三唑-3,5-二基、二-1,2,4-三唑并[4,5-f:4,5-q]-5,6,12,13-四氢-5,12-二氮杂二苯并[a,h]蒽-5,13-二基、9-烷基咔唑-2,7-二基、6,12-二烷基吲哚并[2,3-b]咔唑-2,8-二基、苯并[1,2-b:4,5-b’]二噻吩-2,6-二基、苯并[1,2-b:5,4-b’]二噻吩-2,6-二基、[1]苯并噻吩并[3,2-b][1]苯并噻吩-2,7-二基、苯并[1,2-d:4,5-d’]二噁唑-2,6-二基、苯并[1,2-d:5,4-d’]二噁唑-2,6-二基、5,5-二氧杂二苯并噻吩-3,7-二基或6,12-二烷基-5,5-11,11-四氧杂苯并[1,2-b:4,5-b’]二[1]苯并噻吩-3,9-二基双自由基,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)或(Ig)的化合物所定义的。
在一个实施方式(例如化合物将用作空穴传输材料的实施方式)中,提供了式Ij的化合物,其中Ar1和Ar2在每次出现时独立地选自包括Ara和键的组中,其中Ara表示包括1个芳族、杂芳族或FL部分,或者2或3个通过单键互连的芳族、杂芳族和/或FL部分的双自由基,并且其中芳族的和杂芳族的部分在每次出现时独立地选自由以下组成的组中:1,4-亚苯基、联苯-4,4’-二基、三联苯-4,4"-二基、萘-1,4-二基、萘-2,6-二基、噻吩-2,5-二基、苝-3,10-二基、芘-2,7-二基、2,2’-二噻吩-5,5’-二基、噻吩并[3,2-b]噻吩-2,5-二基、二噻吩并[3,2-b:2’,3’-d]噻吩-2,6-二基、二苯并噻吩-3,7-二基、苯并[1,2-b:4,5-b’]二[1]苯并噻吩-3,9-二基、9-烷基咔唑-2,7-二基、6,12-二烷基吲哚并[2,3-b]咔唑-2,8-二基、苯并[1,2-b:4,5-b’]二噻吩-2,6-二基、苯并[1,2-b:5,4-b’]二噻吩-2,6-二基、或[1]苯并噻吩并[3,2-b][1]苯并噻吩-2,7-二基双自由基,进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)或(Ig)的化合物所定义的。
在一个实施方式(例如化合物将用作电子传输材料的实施方式)中,提供了式Ik的化合物,其中Ar1和Ar2在每次出现时独立地选自包括Ara和键的组中,其中Ara表示包括1个芳族、杂芳族或FL部分,或者2或3个通过单键互连的芳族、杂芳族和/或FL部分的双自由基,并且其中芳族的和杂芳族的部分在每次出现时独立地选自由1,4-亚苯基、联苯-4,4’-二基、三联苯-4,4"-二基、萘-1,4-二基、萘-2,6-二基、嘧啶-2,5-二基、苝-3,10-二基、芘-2,7-二基、噁唑-2,5-二基、1,3,4-噁二唑-2,5-二基、噁唑并[4,5-d]噁唑-2,5-二基、噁唑并[5,4-d]噁唑-2,5-二基、4-烷基-1,2,4-三唑-3,5-二基、4-芳基-1,2,4-三唑-3,5-二基、4-苯基-1,2,4-三唑-3,5-二基、4-(对叔丁基苯基)-1,2,4-三唑-3,5-二基、二-1,2,4-三唑并[4,5-f:4,5-q]-5,6,12,13-四氢-5,12-二氮杂二苯并[a,h]蒽-5,13-二基、咪唑并[4,5-d]咪唑-2,5-二基、苯并[1,2-d:4,5-d’]二噁唑-2,6-二基、苯并[1,2-d:5,4-d’]二噁唑-2,6-二基、5,5-二氧杂二苯并噻吩-3,7-二基或6,12-二烷基-5,5-11,11-四氧杂苯并[1,2-b:4,5-b’]二[1]苯并噻吩-3,9-二基双自由基组成的组中,并且进一步地,其中D、B1、B2、B3、S1、S1a、S2、S2a、S3、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)或(Ig)的化合物所定义的。
在一个实施方式中,提供了式IL的化合物,其中S1、S1a、S2和S2a在每次出现时独立地选自直链或支链的非手性C5-C14烷基基团,可选地,其中1、2、3、4或5个亚甲基基团由氧原子代替,条件是不存在缩醛、缩酮或过氧化物,通过键或醚、酯、碳酸酯、硫醚、胺或酰胺连接基连接至A并通过键或醚、酯、碳酸酯、硫醚、胺或酰胺连接基连接至D、B1、B2、B3或S3,这通过D的性质确定,进一步地,其中D、B1、B2、B3、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)或(Ik)的化合物所定义的。
在一个实施方式中,提供了式Im的化合物,其中S1、S1a、S2和S2a在每次出现时独立地选自直链或支链的非手性C5-C14烷基基团,可选地其中1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮或过氧化物,通过键或醚、酯或碳酸酯连接基连接至A并且通过键、醚、酯或碳酸酯连接基连接至D、B1、B2、B3或S3,这通过D的性质确定,进一步地,其中D、B1、B2、B3、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)或(Ik)的化合物所定义的。
在一个实施方式中,提供了式In的化合物,其中S1、S1a、S2和S2a在每次出现时独立地选自直链或支链的非手性C7-C12烷基基团,可选地其中1、2、3或4个亚甲基基团被氧原子代替,条件是不产生缩醛、缩酮或过氧化物,通过键或醚、酯、碳酸酯、硫醚、胺或酰胺连接基连接至A并且通过键、醚、酯、碳酸酯、硫醚、胺或酰胺连接基连接至D、B1、B2、B3或S3,这通过D的性质确定,进一步地,其中D、B1、B2、B3、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)或(Ik)的化合物所定义的。
在一个实施方式中,提供了式Io的化合物,其中S1、S1a、S2和S2a在每次出现时独立地选自直链或支链的非手性C7-C12烷基基团,可选地其中1、2、3或4个亚甲基基团替代成氧原子,条件是不产生缩醛、缩酮或过氧化物,通过键或醚、酯或碳酸酯连接基连接至A并且通过键、醚、酯或碳酸酯连接基连接至D、B1、B2、B3或S3,这通过D的性质确定,进一步地,其中D、B1、B2、B3、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)或(Ik)的化合物所定义的。
在一个实施方式中,提供了式Ip的化合物,其中当B1、B2、B3和D表示交联基团时,在每次出现时它独立地表示辐射活化的交联基团,可选地光可聚合的交联基团,进一步地,其中S1、S1a、S2、S2a、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)或(Io)的化合物所定义的。
在一个实施方式中,提供了式Iq的化合物,其中当B1、B2、B3和D表示交联基团时,在每次出现时它选自包括烯烃交联基团的组中,进一步地,其中S1、S1a、S2、S2a、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)或(Io)的化合物所定义的。
在一个实施方式中,提供了式Ir的化合物,其中当B1、B2、B3和D表示交联基团时,在每次出现时它独立地表示富电子或贫电子烯烃交联基团,进一步地,其中S1、S1a、S2、S2a、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)或(Io)的化合物所定义的。
在一个实施方式中,提供了式Is的化合物,其中当B1、B2、B3和D表示交联基团时,在每次出现时它独立地表示光可聚合的烯烃交联基团,进一步地,其中S1、S1a、S2、S2a、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)或(Io)的化合物所定义的。
在一个实施方式中,提供了式It的化合物,其中当B1、B2、B3和D表示交联基团时,在每次出现时它独立地表示选自由直链和环状α,β-不饱和酯、α,β-不饱和酰胺、乙烯基醚、非共轭二烯所组成的组中的烯烃交联基团,进一步地,其中S1、S1a、S2、S2a、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、或(Io)的化合物所定义的。
在一个实施方式中,提供了式Iu的化合物,其中当B1、B2、B3和D表示交联基团时,在每次出现时它独立地表示选自由甲基丙烯酸酯基、乙基丙烯酸酯基、乙基马来酸酯基、乙基富马酸酯基、N-马来酰胺基、乙烯氧基、烷基乙烯氧基、乙烯基马来酸酯基、乙烯基富马酸酯基、N-(2-乙烯氧基马来酰胺基)、1,4-戊二烯-3-基和1,4-环己二烯基基团组成的组中的烯烃交联基团,进一步地,其中S1、S1a、S2、S2a、S3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、或(Io)的化合物所定义的。
在一个实施方式中,提供了式Iv的化合物,其中S3表示C1-C20烷基基团、C1-C20卤代烷基基团、C3-C8环烷基基团、C6-C16芳基基团或C4-C15杂芳基基团或由1、2、3、4或5个每个独立地通过键、醚连接基或酯连接基连接的C1-C20烷基、C1-C20卤代烷基、C3-C8环烷基、C6-C16芳基和/或C4-C15杂芳基部分构成的链并且其中S1、S1a、S2、S2a,D、B1、B2、B3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)或(Iu)的化合物所定义的。
在一个实施方式中,提供了式Iw的化合物,其中S3表示C1-C14烷基基团、C1-C14卤代烷基基团、C5-C6环烷基基团、C6-C14芳基基团或由1、2、3、4或5个每个独立地通过键、醚连接基或酯连接基连接的C1-C14烷基、C1-C14卤代烷基、C3-C8环烷基和/或C6-C14芳基部分构成的链,其中S1、S1a、S2、S2a,D、B1、B2、B3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)或(Iu)的化合物所定义的。
在一个实施方式中,提供了式Ix的化合物,其中S3表示C2-C10烷基基团、C2-C10卤代烷基基团、C5-C6环烷基基团、C6-C14芳基基团或由1、2、3、4或5个每个独立地通过键、醚连接基或酯连接基连接的C1-C14烷基、C2-C10卤代烷基、C3-C8环烷基和/或C6-C14芳基部分构成的链,其中S1、S1a、S2、S2a,D、B1、B2、B3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)或(Iu)的化合物所定义的。
在一个实施方式中,提供了式Iy的化合物,其中S3表示由通过键、醚连接基或酯连接基相互连接的三个C2-C12烷基基团和两个C6-C16芳基基团构成的基团;其中每个C6-C16芳基基团i)通过C2-C12烷基基团连接至第二个C6-C16芳基基团,ii)通过C2-C12烷基基团连接至交联剂B2或B3和iii)直接连接至S1和S1a,并且其中S1、S1a、S2、S2a、D、B1、B2、B3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)或(Iu)的化合物所定义的。
在一个实施方式中,提供了式Iz的化合物,其中S3表示由通过键、醚连接基或酯连接基相互连接的四个连接至中心C6-C16芳基基团的C2-C12烷基基团构成的基团,其中每个独立的C2-C12烷基基团未连接至中心C6-C16芳基基团的端中止于B2、B3、S1和S1a的连接中,并且其中S1、S1a、S2、S2a,D、B1、B2、B3、Ar1、Ar2、FL、R和n如上对于式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)或(Iu)的化合物所定义的。
在一个实施方式中,提供了通过交联多个式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)、(Iu)、(Iv)、(Iw)、(Ix)、(Iy)或(Iz)的单体而形成的网状聚合物。
在一个实施方式中,提供了用于制作OLED器件的具有根据式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)、(Iu)、(Iv)、(Iw)、(Ix)、(Iy)或(Iz)的结构的化合物。
在一个实施方式中,提供了具有发光层的OLED器件,该发光层含有下式的2,7-二取代的9,9-氟烷基芴双自由基,
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地,其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了具有发光层的OLED器件,发光层含有i)多个式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)、(Iu)、(Iv)、(Iw)、(Ix)、(Iy)或(Iz)的化合物;或ii)通过交联多个式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)、(Iu)、(Iv)、(Iw)、(Ix)、(Iy)或(Iz)的单体形成(或可获得)的网状聚合物。
在一个实施方式中,提供了具有聚合物发光层的OLED器件,该聚合物发光层含有以下式的2,7-二取代的9,9-氟烷基芴双自由基:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物,进一步地,其中聚合物发光层通过将多个单体暴露于辐射而形成(或可获得),可选地其中辐射是紫外光。
在一个实施方式中,提供了包含聚合物发光层的OLED器件,该聚合物发光层通过将多个式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)、(Iu)、(Iv)、(Iw)、(Ix)、(Iy)或(Iz)的单体暴露于辐射而形成(或可获得)的,可选地,其中辐射是紫外光。
在一个实施方式中,提供了具有电荷传输层的器件,该电荷传输层含有以下式的2,7-二取代的9,9-氟烷基芴双自由基:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,本文提供了包含电荷传输层的器件,该电荷传输层含有i)多个式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)、(Iu)、(Iv)、(Iw)、(Ix)、(Iy)或(Iz)的化合物;或ii)通过交联多个式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)、(Iu)、(Iv)、(Iw)、(Ix)、(Iy)或(Iz)的单体形成(或可获得)的网状聚合物。
在一个实施方式中,提供了具有聚合物电荷传输层的器件,该聚合物电荷传输层含有下式的2,7-二取代的9,9-氟烷基芴双自由基:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物,进一步地,其中聚合物发光层通过将单体暴露于辐射形成(或可获得),可选地其中辐射是紫外光。
在一个实施方式中,提供了含有聚合物电荷传输层的器件,通过将多个式(I)、(Ia)、(Ib)、(Ic)、(Id)、(Ie)、(If)、(Ig)、(Ih)、(Ii)、(Ij)、(Ik)、(IL)、(Im)、(In)、(Io)、(Ip)、(Iq)、(Ir)、(Is)、(It)、(Iu)、(Iv)、(Iw)、(Ix)、(Iy)或(Iz)的单体暴露于辐射而形成(或可获得)的,可选地,其中辐射是紫外光。
在一个实施方式中,提供了包含电荷传输层的器件,该电荷传输层含有下式的2,7-二取代的9,9-氟烷基芴双自由基:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了包括在空穴传输层和电子传输层之间的界面的器件,其中任一或两个所述层都是包含下式的2,7-二取代的9,9-氟烷基芴双自由基的层:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了如上所述的包括在空穴传输层和电子传输层之间的界面的器件,进一步地,其中该器件是光伏器件或薄膜晶体管(TFT)器件。
在一个实施方式中,提供了包含多个层的器件,所述层含有下式的2,7-二取代的9,9-氟烷基芴双自由基:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物,进一步地其中每个层通过反复按序沉积和原位聚合过程形成(或可获得)。
在一个实施方式中,提供了包含多种图案化结构的器件,该图案化结构通过将多个含有下式的2,7-二取代的9,9-氟烷基芴双自由基的材料层暴露于辐射如紫外光的图案化区域以使得该材料层聚合以及随后洗掉未暴露的和未聚合的材料而产生(或可获得):
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了包含两种或多种前述图案化结构的器件,所述结构由性质上是电致发光的材料构成,其中由一种图案化结构发射的电致发光的波长不同于由至少一种其它图案化结构发射的电致发光的波长。
在一个实施方式中,提供了多色点阵显示器,包括大量多色像素,每个像素包括重述图案化结构的一种或多种。
在一个实施方式中,提供了包含两种或更多种前述图案化结构的器件,所述结构由性质上是电致发光的材料构成,其中两种或更多种前述图案化结构叠层成堆,进一步地,其中每个堆中的两种或更多种图案化结构的电致发光波长是不同的。
在一个实施方式中,提供了包含聚合的液晶材料的器件,该聚合的液晶材料包括下式的2,7-二取代的9,9-氟烷基芴双自由基:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了包含玻璃的器件,该玻璃通过冷却包含下式的2,7-二取代的9,9-氟烷基芴双自由基的液晶材料形成(或可获得):
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了包含聚合向列液晶材料的器件,该材料包括下式的2,7-二取代的9,9-氟烷基芴双自由基:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了包含聚合物基质的器件,该聚合物基质通过将包含含下式的2,7-二取代的9,9-氟烷基芴双自由基的分子的液晶流体暴露于辐射而形成(或可获得),
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物,可选地其中辐射是紫外光。
在一个实施方式中,包含如上所述的聚合物基质的OLED器件的特征在于该基质包括已经适当锁定的向列液晶结构。
在一个实施方式中,提供了包含发光层的OLED器件,该发光层包括具有均匀对齐的液晶结构的材料,该材料包括包含下式的2,7-二取代的9,9-氟烷基芴双自由基的分子使得所述发光层发射线性偏振光:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了用于形成包含多种图案化结构的器件的方法,所述方法包括将多个包括下式的2,7-二取代的9,9-氟烷基芴双自由基的材料层暴露于辐射如紫外光的图案化区域以促使所述材料层聚合和随后洗掉未暴露的和未聚合的材料,
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了通过将均匀对齐的液晶流体的层或通过冷却包含含有下式的2,7-二取代的9,9-氟烷基芴双自由基的可交联分子的均匀对齐的液晶流体而形成(或可获得)的玻璃暴露于辐射如紫外光的图案化区域,和随后洗掉未暴露的和未聚合的材料而制作(或可获得)的结构,
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物,其中所述暴露于辐射导致所述暴露的材料层发生聚合。
在一个实施方式中,提供了偏振光发射结构,通过将均匀对齐的液晶流体的层或通过冷却包含含有下式的2,7-二取代的9,9-氟烷基芴双自由基的可交联分子的均匀对齐的液晶流体而形成(或可获得)的玻璃暴露于辐射,可选地紫外光的图案化区域,并随后洗掉未暴露的和未聚合的材料而制作(或可获得),
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物,其中所述暴露于辐射导致所述暴露的材料层发生聚合。
在一个实施方式中,提供了包括两种或更多种以上所述的图案化偏振光发射结构的器件,其中至少第一偏振光发射结构具有与至少第二偏振光发射结构的光发射偏振轴不对齐的光发射偏振轴。
在一个实施方式中,提供了3D显示器,其通过按序沉积均匀对齐的液晶流体的对齐层或通过冷却包含含有下式的2,7-二取代的9,9-氟烷基芴双自由基的可交联分子的均匀对齐的液晶流体而形成(或可获得)的玻璃,依次按序聚合每层的图案化区域,并依次按序漂洗掉每层未聚合区域,以便提供发光结构而使液晶对齐和由此的每个各个层的发光偏振轴处于与各个毗邻层的发光偏振轴的方向不同的方向上而产生(或可获得),
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了包括聚合物的器件,该聚合物包含2,7-二取代的9,9-氟烷基芴重复单元:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了包括发光聚合物的OLED器件,该发光聚合物包含2,7-二取代的9,9-氟烷基芴重复单元:
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物。
在一个实施方式中,提供了包含含有2,7-二取代的9,9-氟烷基芴重复单元的聚合物的OLED器件
其中R表示直链或支链的非手性C1-C14烷基、C1-C14卤代烷基或C2-C14烯基基团,可选地其中R的1、2、3、4或5个亚甲基基团被氧原子代替,条件是不存在缩醛、缩酮、过氧化物或乙烯基醚,和这些基团的部分或完全氟化的衍生物并且其中该聚合物用作具有液晶结构的宿主材料中的发光掺杂剂。
在一个方面中,本发明涉及化合物
其中R在每种情况下是直链或支链的非手性C1-C14烷基、C1-C14卤代烷基、C1-C14氟烷基、C2-C14烯基基团,可选地其中1、2、3、4或5个CH2基团被氧取代,条件是在R基团中不存在缩醛、缩酮、过氧化物或乙烯基醚。在优选的实施方式中,R在每种情况下是直链或支链的非手性C2-10烷基基团。在优选的实施方式中,R在每种情况下是直链C2-10烷基基团。在这些化合物优选的实施方式中,R基团是相同的。
在一个方面中,本发明涉及用于制备结构B1-S2-A-S1-S3(B2)(B3)-S1a-A-S2a-B1a的物质的方法,涉及用以下结构的化合物烷基化酚氧(酚羟基氧,phenolic oxygen)的步骤,
其中,R1和R2独立地是C1-12烷基、C6-10芳基或C5-9杂芳基;
L1和L2独立地选自离去基团,可选地选自Cl、Br、I、O-甲苯磺酰基、O-甲磺酰基或O-三氟甲磺酰基;和
m和n是1至10的整数。
在一个方面中,本发明涉及二乙基-2,5-二(溴己基)氧对苯二甲酸酯,二乙基-2,5-二(氯己基)氧对苯二甲酸酯,二乙基-2,5-二(碘己基)氧对苯二甲酸酯和具有C1-12烷基、C6-10芳基或C5-9杂芳基酯基团的类似化合物。在一个方面中,本发明涉及二乙基-2,5-二(溴己基)氧对苯二甲酸酯、二乙基-2,5-二(氯己基)氧对苯二甲酸酯和二乙基-2,5-二(碘己基)氧对苯二甲酸酯以及具有甲基、丙基、丁基、戊基和己基酯基团的类似化合物作为用于制作具有对于以上化合物(I)、(Iv)、(Iw)、(Ix)和(Iz)所定义的通式结构S3的可交联物质的中间体的用途。
具体实施方式
根据本发明的一方面,提供了D-S1-A-S2-B1的化合物,其中基团D、B1、S1、S2和A是本文中定义的化学基团。A表示式-Ar1-(FL-Ar2)n-的基团。体系的组分部分通过共价键彼此连接。
该基团也可以表示为式1的化合物
使得可以更好地理解本发明,构成基团的性质以及它们的功能定义于本文中。
Ar1-(FL-Ar2)n-
本发明的化合物包含形成化合物的“基本线性的”,或“梭板样”的芳族核芯的-Ar1-(FL-Ar2)n-基团,缩写为A。在该结构中,FL是以下结构的芴双自由基,其在C-2和C-7位通过共价键并入链中
在-Ar1-(FL-Ar2)n-中的整数n为1至8。总计,每个-Ar1-(FL-Ar2)n-基团包含1至8个FL基团。在某些优选方面中,每个-Ar1-(FL-Ar2)n-基团的FL数目选自1至6、1至3、3至6、或5、或6。
在本发明的化合物适合用作发光器的优选的方面中,每个-Ar1-(FL-Ar2)n-基团包含3至6个FL基团。这是因为较低的n值导致具有较低的发光效率的分子。在本发明的化合物适合用作空穴传输器、电子传输器或发光掺杂剂的宿主的实施方式中,具有1至3个FL基团的-Ar1-(FL-Ar2)n-基团是优选的,因为其易于合成、熔点较低且溶解度较高。
在这种-Ar1-(FL-Ar2)n-或A结构中,当n大于1时每个单独的FL的R基团是相同的,因为该物质的多个异构体具有使纯化具有挑战性的潜力。对于不同FL基团中具有不同R基团而n=1的材料,可能是有利的,因为这可以导致熔点降低。R基团选自直链或支链的非手性C1-C14烷基、C1-C14卤代烷基、C1-C14氟烷基、C2-C14烯基基团,可选地其中1、2、3、4或5个CH2基团被氧代替,使得不存在缩醛、缩酮、过氧化物或乙烯基醚。换句话说,如果多于一个亚甲基基团被氧原子代替,则它将在链中通过至少3个共价键与下一个氧原子分开。当R基团是烯基基团时,优选烯烃是端烯烃,因为产物的粘度能够通过将端烯烃引入R基团中而降低。
Ar1和Ar2在每次出现时独立地选自包括Ara和键的组中。Ara表示包括1个芳族、杂芳族或FL部分,或者2、3、4或5个通过单键互连的芳族、杂芳族和/或FL部分的双自由基。
整个Ar1-(FL-Ar2)n链是“基本上线性的”,并缺乏会破坏其线性度、其与相邻分子对齐并有利于液体结晶度的能力的显著支化。链的某些成分可以从线性结构突出。例如,当Ar1是萘-1,4-二基时,碳5至8从链的侧面突出,即使萘基结构整合到链中。可以理解的是,虽然链作为整体描述为线性的,但连接链的组成部件的化学键的性质决定了链中的所有化学键将并不会准确对齐。只要材料分子的分子核芯的骨架中的任何弯曲不会破坏材料的液晶性质或至少其通过液晶分子对齐的能力,那么所述完全是允许的。同样,也允许保留材料液晶性质的支化。
R
R基团选自直链或支链的非手性C1-C14烷基、C1-C14卤代烷基、C1-C14氟烷基、C2-C14烯基基团,可选地其中1、2、3、4或5个亚甲基(CH2)基团被氧代替,使得不存在缩醛、缩酮、过氧化物或乙烯基醚。换句话说,如果多于一个亚甲基基团被氧原子代替,则它将会被至少三个共价键与链上的另一个氧原子分开并且没有氧原子通过单键连接至碳碳双键。在一些方面中,每个单独的芴的R基团是相同的。在另一方面中,链上的每个芴上的R基团是相同的。
在优选的方面中,每个FL部分的R基团是相同的并选自由直链或支链的非手性C2-C10烷基、C2-C10卤代烷基、C2-C10氟烷基、C2-C10烯基基团组成的组中,可选地其中1、2或3个CH2基团被氧代替,条件是不存在缩醛、缩酮或过氧化物。
在优选的方面中,每个R基团的每个氢原子可以独立地被氟原子取代。当R基团含有至少一个氟取代基时,其能够称之为氟烷基基团。在优选的方面中,氟烷基基团包含至少一个氢原子。卤代烷基基团包括被Fl、Cl、Br和I取代的基团。优选的卤代烷基基团是氟烷基、氯烷基和氟氯烷基基团。
在优选的方面中,R基团是烯基基团。烯基基团含有碳碳双键。优选的烯基基团仅含有一个碳碳双键。在优选的方面中,在R基团是烯基基团的情况下,它是端碳碳双键。在优选的方面中,端烯烃是式的-CH=CH2。
正如上所述,在某些情况下R基团中的1个或2个或3个或4个或5个亚甲基基团可以被氧原子代替。当这属于这种情况时,R基团是醚或聚醚。亚甲基基团是CH2基团。当两个或更多个亚甲基基团被氧原子代替时,在链上它们之间具有至少两个碳原子。这是因为过氧化物、缩酮、缩醛和乙烯基醚单元潜在地是不稳定的并因此不包括于本发明的化合物的结构中。
因为能够调节化合物的熔点,所以R基团长度的变化是有用的。例如,当需要液晶化合物时,可能有利的是在链中使用具有2至14个碳和氧原子的R基团,因为C1烃基衍生物经常表现出升高的熔点。在优选的方面中,R基团在链中含有2至10个碳和氧原子。
将氧原子引入到R基团中能够有利地用于调节化合物其玻璃化转变的温度并且这对于需要玻璃态材料时的应用能够是一个优点。
引入碳碳双键能够有利地用于降低材料的粘度,并且这对于将会进行溶液加工的化合物是特别有意义的。在链末端引入碳碳双键对于调节材料的粘度是特别有利的。
对于电子器件中需要高纯度材料的应用,则烃基基团中任何不对称中心的存在都是不利的。出于这个原因,在本发明R基团被支化的化合物中,支化不要引入手性中心。这种非手性支化有利地规避了与能够与化学物质的非对映体混合物有关的纯化的问题。基团R中引入支化能够有利地用于调节本发明的化合物的熔点。
具有完全引入的R基团的FL结构的所选实例如下所示。
以下结构的化合物
由于其合成效用是用于合成含有这些氟烷基芴衍生物发射体核芯的化合物的优选中间体。
Ar1和Ar2
Ar1和Ar2在每次出现时独立地选自包括Ara和键的组中。Ara表示包括1个芳族、杂芳族或FL部分,或者2、3、4或5个通过单键互连的芳族、杂芳族和/或FL部分的双自由基。双自由基是在化合物的整个结构中共价连接至两个其它部分的基团,以下示出了一些典型的实例,*表示连接的典型位点。
选择的Ara基团的精确性质取决于体系中所需的性质。例如,如果需要发光化合物,则两个、三个、四个、五个或六个相连的FL基团可以出现在结构中。可替换地,高比例的Ara基团,例如,50%或更高,可以是FL基团。
由Ara包含的组分芳族和杂芳族的基团能够选自可选取代的C6-C16芳族基团和C4-C12杂芳族基团的组中。可选的取代基可以选自可选地非手性支化的C1-C10烷基或醚基团的组中。
适用作本发明材料中的Ara结构单元的芳族双自由基包括,但不限于,1,4-亚苯基、联苯-4,4’-二基、三联苯-4,4"-二基、萘-1,4-二基、萘-2,6-二基、噻吩-2,5-二基、嘧啶-2,5-二基、吡啶-2,5-二基、苝-3,10-二基、芘-2,7-二基、2,2’-二噻吩-5,5’-二基、噁唑-2,5-二基、1,3,4-噁二唑-2,5-二基、噻吩并[3,2-b]噻吩-2,5-二基、二噻吩并[3,2-b:2’,3’-d]噻吩-2,6-二基、二苯并噻吩-3,7-二基、苯并[1,2-b:4,5-b’]二[1]苯并噻吩-3,9-二基、噻唑并[5,4-d]噻唑-2,5-二基、噁唑并[5,4-d]噁唑-2,5-二基、噻唑并[5,4-d]噁唑-2,5-二基、噻唑并[4,5-d]噻唑-2,5-二基、噁唑并[4,5-d]噁唑-2,5-二基、噻唑并[4,5-d]噁唑-2,5-二基、2,1,3-苯并噻二唑-4,7-二基、4-噻吩-2-基-2,1,3-苯并噻唑-7,5’-二基、4,7-二噻吩-2-基-2,1,3-苯并噻唑-5’,5”-二基、咪唑并[4,5-d]咪唑-2,5-二基、4-烷基-1,2,4-三唑-3,5-二基、4-芳基-1,2,4-三唑-3,5-二基、4-苯基-1,2,4-三唑-3,5-二基、4-(对叔丁基苯基)-1,2,4-三唑-3,5-二基、二-1,2,4-三唑并[4,5-f:4,5-q]-5,6,12,13-四氢-5,12-二氮杂二苯并[a,h]蒽-5,13-二基、9-烷基咔唑-2,7-二基、6,12-二烷基吲哚并[2,3-b]咔唑-2,8-二基、苯并[1,2-b:4,5-b’]二噻吩-2,6-二基、苯并[1,2-b:5,4-b’]二噻吩-2,6-二基、[1]苯并噻吩并[3,2-b][1]苯并噻吩-2,7-二基、苯并[1,2-d:4,5-d’]二噁唑-2,6-二基、苯并[1,2-d:5,4-d’]二噁唑-2,6-二基、5,5-二氧杂二苯并噻吩-3,7-二基或6,12-二烷基-5,5-11,11-四氧杂苯并[1,2-b:4,5-b’]二[1]苯并噻吩-3,9-二基双自由基,如果需要空穴传输性能,则含吲哚和噻吩的部分如以下所示的那些可能是优选的,*表示连接的典型位点。
如果需要电子传输性能,则含噁唑的部分如以下所示的那些可能是优选的,*表示连接的典型位点。
在优选的方面中,该化合物是发射体材料而n为1至6。在进一步优选的方面中,n为3至6。在优选的方面中,n为5或6。在优选的方面中,n为5。在进一步优选的方面中,n为6。更长的分子是优选的,因为器件的能量效率随着分子的芳族核芯的长度增加而提高。由发光衰减时间测量的证据表明,能量效率的增加是由于较长的分子核芯中出现,但不太可能在较短的分子核芯中发生的三重态-三重激发子湮灭。此外,在能量效率上的小幅增加能够通过进一步增长分子核芯至n=7或8而实现,但效率的提高必须与合成这类分子的成本增加相平衡。
本发明进一步优选的方面是本发明包含分子核芯结构A的发射体材料用FL单元终止。在电荷传输或宿主材料分子中,用Ar单元如联苯-4,4’-二基终止分子核芯可能是有利的,因为材料的液晶性能能够提高而电荷载流子传输能够通过端Ar基团之间的π-π相互作用介导的强分子间相互作用增强。然而,在发射体材料的情况下,这些相同的相互作用能够导致激发子能量淬火并且从而在9-位置上具有相对大体积的取代基的端FL基团是优选的。
柔性连接基团S1、S1a、S2和S2a
当D表示可交联基团时,本发明的化合物包含两个柔性连接基团S1和S2。正如本文中的讨论,D也能够表示含有其它类似于S1和S2基团的柔性连接基团S1a和S2a的更复杂结构。在每次出现时,S1、S1a、S2和S2a独立地选自直链或支链的非手性C5-C14烷基基团,可选地其中1、2、3、4或5个亚甲基基团被氧原子代替,条件是R不含有过氧化物、缩酮或缩醛基团,通过键或醚、酯、碳酸酯、硫醚、胺或酰胺连接基连接至A并通过键或醚、酯、碳酸酯、硫醚、胺或酰胺连接基连接至D、B1、B2、B3或S3,这通过D的性质确定。
柔性连接基团S1、S1a、S2和S2a用于将A中的荧光团体系与可交联的基团分开。当材料交联至网状聚合物基质时,柔性连接基机械上和电子上将荧光团与聚合物基质分隔开。因此,当材料交联至聚合物基质时,柔性连接基用于降低非发光激发子的淬灭由此有利于有效发光。
D、B1、B2和B3
D表示可交联基团或,当B1表示氢时,D表示-B2-S3-B3-S1a-A-S2a-B1a、-S3(B2)-B3-S1a-A-S2a-B1a、-S3(B2)(B3)-S1a-A-S2a-B1a、-S3(B2)(B3)或其中链左端上的破折号表示连接至S1的连接点的可交联基团。B1、B2和B3各自独立地表示交联基团或氢。
因此本发明的化合物包含交联基团,并且当交联时,形成网状聚合物。这是因为优选的交联基团与两个其它交联基团反应而产生链反应和聚合物基质。
在优选的方面中,交联基团选自乙烯型(烯型,ethylenic)、二烯型、硫醇和氧杂环丁烷型可交联基团的组中。乙烯型可交联基团是含有碳碳双键的可交联基团。在优选的方面中,所有交联基团独立地表示乙烯型交联基团。有利的乙烯型交联基团包括富电子和贫电子乙烯型交联基团。
在优选的方面中,可交联的基团在暴露于辐射时经受交联反应。在优选的方面中,可交联的基团在暴露于紫外(UV)光时经受交联反应。
优选的交联基团的实例是直链和环状的α,β-不饱和酯、α,β-不饱和酰胺、乙烯基醚和非共轭的二烯交联基团。有利的交联基团因此包括甲基丙烯酸酯基、乙基丙烯酸酯基、乙基马来酸酯基、乙基富马酸酯基、N-马来酰胺基、乙烯氧基、烷基乙烯氧基、乙烯基马来酸酯基、乙烯基富马酸酯基、N-(2-乙烯氧基马来酰胺基)、1,4-戊二烯-3-基和1,4-环己二烯基基团。
在优选的方面中,交联基团是富电子乙烯型可交联基团。富电子乙烯型可交联基团含有用一个或多个供电子基团取代的乙烯基团。供电子基团能够包含杂原子如O、N或S。在优选的方面中,富电子可交联基团是乙烯氧基基团。其它供电子基团取代的交联基团是1-烯基醚如丙烯-1-氧基基团和丁烯-1-氧基基团;环状乙烯基醚如环己烯-1-氧基和环戊烯-1-氧基;双环乙烯基醚如2-降冰片烯-2-氧基基团和其中乙烯基醚官能度通过干预烃基结构(intervening hydrocarbyl structure)连接至柔性连接基或间隔基(S1、S1a、S2、S2a或S3)的基团如4-乙烯氧基苯氧基和2-乙烯氧基乙基基团。
在优选的方面中,交联基团是贫电子乙烯型可交联基团。贫电子的乙烯型可交联基团含有用一个或多个吸电子基团取代的乙烯基团。吸电子基团可以包含羰基基团并可以例如是酯或酰胺。在优选的方面中,贫电子可交联基团包含单烷基马来酸酯基团、单烷基富马酸酯基团、单芳基马来酸酯基团、单芳基富马酸酯基团或马来酰胺基团。贫电子交联基团的其它实例有4,4,4-三氟巴豆酸酯基团、Z-4,4,4-三氟丁烯酸酯基团、3-三氟甲基-4,4,4-三氟巴豆酸酯基团、Z-和E-3-氰基丙烯酸酯、Z-和E-3-氰基甲基丙烯酸酯、单烷基环己烯-1,2-二甲酸酯和单烷基环戊烯-1,2-二甲酸酯。
正如上所述,D表示可交联基团或,当B1表示氢时,D能够表示-B2-S3-B3-S1a-A-S2a-B1a、-S3(B2)-B3-S1a-A-S2a-B1a、-S3(B2)(B3)-S1a-A-S2a-B1a或-S3(B2)(B3)或链左端上的破折号表示连接至S1的连接点的交联基团。
因此,在一个方面中,D是结构-B2-S3-B3-S1a-A-S2a-B1a以及D的所有成分连接在线性链内。这种排布的一个实例,其中为了举例说明的目的,可交联基团B2和B3是单甲基马来酸酯基团以及B1和B1a表示氢,整个结构是以下所示的“1型”S3间隔基。
在第二方面中,D是结构-S3(B2)-B3-S1-A-S2a-B1a以及一个可交联基团,B2,形成从S3支化并连接至S3的侧链。这种排布的一个实例,其中为了举例说明的目的,可交联基团B2和B3是单甲基马来酸酯基团以及B1和B1a表示氢,整体结构是以下所示的“2型”S3间隔基。
在第三方面中,D是结构S3(B2)(B3)-S1-A-S2a-B1a,结构中的两个S1基团都通过两个可交联基团连接至其并由其突出的连接基S3桥接。这种排布的一个实例,其中出于举例说明的目的,可交联基团B2和B3是单甲基马来酸酯基团以及B1a和B1表示氢,整个结构是以下所示的“3型”S3间隔基。
在第四方面中,D是结构-S3(B2)(B3)。在这种结构中,间隔基团S3用两个交联基团修饰。这种排布的一个实例,其中出于举例说明的目的,可交联基团B2和B3是单甲基马来酸酯基团以及B1表示氢,整个结构是如下所示的“4型”S3间隔基。
S3
D表示可交联基团或,当B1表示氢时,D能够表示-B2-S3-B3-S1a-A-S2a-B1a、-S3(B2)-B3-S1a-A-S2a-B1a、-S3(B2)(B3)-S1a-A-S2a-B1a、或-S3(B2)(B3)或其中链左端上的破折号表示连接至S1的连接点并且其中B1a表示氢的交联基团。在这种结构中存在另外的间隔基S3。
S3表示可以是刚性的或柔性的非发色团间隔基团。S3可以包括C1-C20烷基基团、C1-C20卤代烷基基团、C3-C8环烷基基团、C6-C16芳基基团或C4-C15杂芳基基团或由1、2、3、4或5个各自独立地通过键、醚连接基或酯连接基连接的C1-C20烷基、C1-C20卤代烷基、C3-C8环烷基、C6-C16芳基和/或C4-C15杂芳基部分构成的链。S3通过键、醚、酯或碳酸酯连接基连接至B2和/或B3。
间隔基S3优选的实例包括C2-C12烷基基团、C3-C8环烷基基团或C6-C16芳基基团或由1、2、3、4或5个各自独立地通过键、醚连接基或酯连接基连接的C1-C20烷基、C1-C20卤代烷基、C3-C8环烷基、C6-C16芳基和/或C4-C15杂芳基部分构成的链。
为清楚起见,所提供的具有B2和B3交联基团的1型S3间隔基团的实例如下所示的(波浪线表示至S1和S1a的连接点):
为清楚起见,提供的具有B2和B3交联基团的2型S3间隔基团的实例有(波浪线表示至其它链组分的连接点):
为清楚起见,所提供的具有B2和B3交联基团的3型S3间隔基团有(波浪线表示至其它链组分的连接点):
X、Y和Z独立地选自键、醚(O)或酯连接基(-CO2),a、b、c、d和e是提供C1至C20的连接基(烷基,卤代烷基)的整数。
3型S3间隔基团是特别有意义的,因为它是将这些基团引入到整个B1-S2-A-S1-S3(B2)(B3)-S1a-A-S2a-B1a结构中的化学中间体。3型S3间隔基团的优选实例包括C2-C12烷基基团、C3-C8环烷基基团或C6-C16芳基基团或由1、2、3、4或5个各自独立地通过键、醚连接基或酯连接基连接的C1-C20烷基、C1-C20卤代烷基、C3-C8环烷基、C6-C16芳基和/或C4-C15杂芳基部分构成的链。在优选的实例中,3型S3间隔基团包括C2-C12烷基基团或C6-C16芳基基团或由1、2、3、4或5个各自独立地通过键、醚连接基或酯连接基连接的C2-C12烷基基团和/或C6-C16芳基基团构成的链。在优选的实例中,3型S3间隔基团包含3、4或5个各自独立地通过键、醚连接基或酯连接基连接的C2-C12烷基基团和/或C6-C16芳基基团。
在优选的实例中,3型S3间隔基团包括5个选自各自独立地通过键、醚连接基或酯连接基连接的C2-C12烷基和C6-C16芳基基团的基团。在3型S3间隔基团中,优选交联基团B2和B3连接至C3-C12烷基基团,可选地连接至C4-C10烷基基团,因为这种结构会提供更大的结构柔性并有助于B2和B3与相邻分子中存在的交联基团的可能的交联反应。这提供了温和条件下进行交联的潜力并最小化可能的降解。
在优选的实例中,3型S3间隔基团包含4个通过键、醚连接基或酯连接基连接至中心C6-C16芳基基团的C2-C12烷基基团。在这种3型S3间隔基团中,各自独立的C2-C12烷基基团的另一端分别终止于对B2、B3、S1和S1a的连接。这种优选种类的3型S3间隔基团的实例如下所示的,其中波浪线表示至其它链组分S1和S1a的连接点,基团X、Y和Z独立地表示键、醚连接基或酯连接基,a和d在每种情况下是2至12的整数以及a和c在每种情况下是1至11的整数。
在进一步的优选实例中,3型S3间隔基团包括三个通过键、醚连接基或酯连接基相互连接的C2-C12烷基基团和两个C6-C16芳基基团。在这种结构中,每个C6-C16芳基基团i)通过C2-C12烷基基团连接至第二C6-C16芳基基团,ii)通过C2-C12烷基基团连接至交联剂B2或B3以及iii)直接连接至S1和S1a。这种优选种类的3型S3间隔基团的实例如下所示的,其中波浪线表示至其它链组分S1和S1a的连接点,基团X、Y和Z独立地表示键、醚连接基或酯连接基,b和d是1至11的整数以及a和c是0至10的整数。
引入优选的3型S3间隔基团的结构的实例如下所示。
为清楚起见,提供的具有B2和B3交联基团的4型S3间隔基团的实例有(波浪线表示至S1的连接点):
因此能够认识到,在某些情况下基团D是如下所示的通式结构类型。
其中B1和B1a表示氢。
本发明的材料的结构D的另外的实例提供如下用于举例说明。R基团中的支化能够用于调节材料的熔点。
材料性能
本发明的材料对于其电荷传输和发光性能都特别有用。因此,本文中描述的材料适用于电子器件,例如有机发光二极管和光伏器件的制作。
本发明的低聚材料的一个有利特性是它们可溶于常见有机溶剂中。这是有意义的,因为低聚物的溶解度性质在器件制作方面相对于例如聚合物材料提供了明显的优势。更详细地说,这些低聚材料可以经由溶液加工方法用于制作器件。概括地说,这包括首先溶解该材料,将该溶液施加于基底上,并随后蒸发以在基底上产生膜涂层。一旦该材料沉积为膜,该材料能够原位聚合。这种聚合可以通过暴露于辐射,例如紫外光而引发,这会导致一个分子的可交联基团与相邻分子中的交联基团交联以形成网状聚合物。沉积的膜的区域能够通过引发的辐射掩蔽掉以获得非交联材料的区域,同时暴露于辐射的区域发生聚合。如果需要,由于交联的材料相对于单体具有可忽略的或降低的溶解度,未暴露的非交联材料能够洗掉而留下交联材料的图案化结构。溶液沉积和聚合的重复循环能够用于产生具有复杂构造的结构。
依次沉积的聚合结构能够按照并排或堆叠/层层叠方式组装。在一个实例中,按照并排方式按序沉积和聚合红、绿和蓝发光材料能够用于为彩色显示器产生像素。在另一实例中,红色、绿色和蓝色发射体材料的叠堆能够用于提供白色光源。在另一实例中,两个或更多个发射体结构能够在叠堆中排布而提供有色光源。
廉价和经济地生产相邻层具有不同的最高占据或最低未占据分子轨道(HOMO和LUMO)能级的多层器件的能力,以及不同电荷载流子迁移率在塑料电子学中是通用用途。例如,使用本发明的材料和方法可以形成p-n结的等效物以及这些可以在二极管、晶体管和光伏器件中找到用途。本发明的材料将进行光刻图案化的倾向容许制作实际上任何尺寸和描述的塑料电子器件的大型阵列。
根据本发明的材料可以混合在一起以形成液晶混合物。这从优化用于预定应用的材料性能的观点可能是非常有利的。例如,本发明的各个化合物可以使液晶具有远低于其熔点(单向转变液晶相)的各向同性液体转变温度。在器件制作应用中,这能够导致材料充分热力学不稳定的玻璃态或过冷液体膜以便导致膜中结晶的危险并随后破坏有用的电子性能。将多组分化合物混合一起能够将所得混合物的熔点降低至低于液晶各向同性液体转变温度或至少充分抑制结晶以便消除这个问题。
与使用本发明材料的混合物相关的另一优点是,它可以允许使用具有其它高度有用的器件应用性能的材料,即使它们具有赋予其作为纯材料不可用的特定属性。例如,制备具有向列液晶结构的发光聚合物膜可能是合乎需要的。本发明的化合物可以是非常高效率的发光材料并具有其他有用的性能,但同时据发现可能具有近晶而不是向列液晶相。通过将合乎需要的化合物溶解至本发明其他化合物具有向列相的混合物中,可能导致产生具有结合了向列相结构的第一高度所需材料的发光性能的混合物。
与使用液晶材料或材料混合物相关的另外的优点是能够形成定向组织化或各向异性的结构。这种定向秩序能够通过,例如通过将沉积的膜暴露于辐射如紫外光使沉积的膜的组分交联而原位固定。
在一个优选的实例中,液晶膜能够沉积于涂覆有取向层如光取向层的基底上。光取向层的组分在暴露于光时形成定向有序的结构。在取向层中的该定向顺序随后能够转移到其表面上形成的沉积的液晶膜中,导致高度有序的器件结构,该器件结构能够通过将沉积的有序膜暴露于辐射如紫外光聚合组分低聚物而原位锁定。
能够利用获得高度有序的器件结构的潜力产生偏振光发射结构,在该偏振光发射结构中发射体核芯在相同方向上对齐并因此在相同方向上发射光。最后,本文中描述的材料的性能提供了通过按序沉积均匀对齐的液晶流体或玻璃的取向层,依次按序聚合每层的图案化区域,和由此随后洗掉每层未聚合区域以提供发光结构,使得液晶对齐和由此的每个相应层的发光偏光轴处于不同的方向上而制作3D-显示器的可能性。
本发明的材料还具有许多另外所需的性能,使它们适用于生产电子器件如OLED。在有机发光器件中,还常需要降低有机发光材料对所发射的光的自吸收。这种自吸收是会发生的,因为有机发光材料的光谱吸收和发射带在各种材料中存在程度或大或小的重叠。这种问题的解决方案是公知的,例如,在染料激光领域中是将发光材料溶解于吸收比发光溶质更短波长的光的宿主中。如果溶液较稀,例如,百分之一到二,则发光溶质的自吸收几乎完全受到抑制。本发明各种化合物的易于互混溶性使得这种类型的溶液制备很容易。本发明的材料因此适用作宿主材料以及发光材料。
在有机发光器件应用中,有必要使激发能量易于从宿主材料传递至溶质发光材料。这是因为电荷载流子(电子和空穴)必须传送通过宿主介质而融合以形成辐射光的激发子(电激发分子轨道状态)。在主要由组分宿主分子构成的混合物中,这种融合和激发子形成将主要发生于宿主分子中。然后激发能量需要从宿主分子传递至发光溶质分子。对于这种能量传递要求宿主材料的光谱发光发射带与发光溶质的吸收带重叠。因此,本发明的重要方面是本发明化合物的混合物的制备,其在构成组分之间具有这种光谱关系。例如,在光谱的蓝色区域中发光的化合物能够充当绿光发射体化合物的宿主。通过5%蓝色发射体化合物在绿色发射体化合物中的溶液的UV引发交联而制备的聚合物膜将表现出比由UV交联纯绿发射体制备的膜相当少的由绿色发射体发射的绿光的自吸收。
为了证明本发明的材料的用途,示例性蓝和绿发射体(“Lomox蓝”和“Lomox绿”如下所示)在没有交联的情况下引入到简单的未优化OLED器件中。在这些器件中,发光层由掺杂到聚(乙烯基咔唑)(PVK),标准宿主材料中的基于选定的氟烷基芴的发射体之一构成。该器件的整个结构是ITO(125nm)/PEDOT:PSS(50nm)/PVK:发射体(70nm)/TPBI(30nm)/LiF(1nm)/Al(100nm)。PEDOT:PSS是标准空穴注入材料,以及TPBI是标准电子传输材料。铟锡氧化物(ITO)用作阳极,而阴极由氟化锂和铝构成。PEDOT:PSS和发光层通过旋涂沉积于预图案化玻璃/ITO基底上,同时随后通过热蒸发沉积TPBI电子传输层和阴极层。
Lomox蓝
标准蓝
Lomox绿
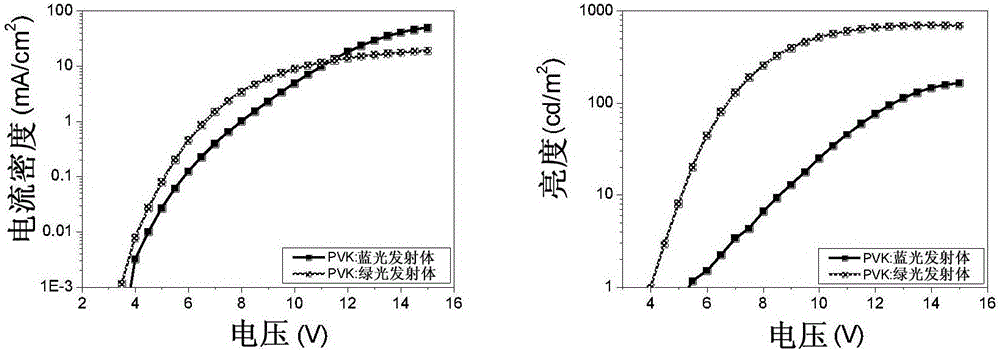
这些器件的电流电压和发光数据如图2中所示,其分别在第一图和在第二图中示出了引入本发明的发射体作为PVK宿主中的掺杂剂的器件的:J-V(左)和L-V(右)数据。
对于本发明具有本文中所示的蓝色发射体的器件,掺杂比为100:5的PVK:发射体,而对于绿器件,该比率为100:15。引入蓝色发射体的器件在15V的电压下达到165cd m-2的峰值亮度,而对于绿色发射体在14伏电压下峰值亮度为700cd m-2。
此外,已经制作出使用蓝色发射体作为绿色发射体的宿主材料的器件,这表明这种材料也能够用作宿主。这种器件引入了PVK层作为空穴传输/电子阻挡中间层,对于整个器件结构则是ITO(125nm)/PEDOT:PSS(50nm)/PVK(30nm)/Lomox蓝色:Lomox绿(50nm)/TPBI(30nm)/LiF(1nm)/Al(100nm)。在所示的器件中Lomox蓝色宿主与Lomox绿色发射体的比率为85:15。
该器件的电流电压和亮度数据如图3中所示。该器件达到的峰值亮度在14.5V电压下为595cd m-2。
该器件的电致发光光谱如图4中所示,示出了来自掺杂剂发射体的主要绿色光发射而同时来自宿主材料的蓝色光发射的较弱峰值能够在350至450nm之间观察到。
使用本发明材料的混合物的另一个优点是,它允许使用反应性液晶元材料的混合物,其中光引发的电子供体/受体相互作用而不是离子或自由基引发用于引发聚合。这可能导致产生比基于甲基丙烯酸酯的体系(在货架寿命方面)稳定得多的反应性液晶元材料,而同时保持了低的UV交联通量。在这些混合物中,反应性液晶元材料中至少一种用富电子交联基团代替,同时至少一种其它组分反应性液晶元材料用贫电子交联基团代替。入射在材料上的紫外辐射促进了某些反应性液晶元分子上的贫电子交联基团转成电子激发态。激发态,贫电子交联基团随后从引发共聚交联反应的其它反应性液晶元分子上的富电子(电子供体)交联基团吸取电子。这种模式的光聚合的描述可以在例如,“Photoinitiated radical polymerization of vinyl ether-maleate systems”,Polymer 38,(9),pp.2229-37(1997);“Co-Polymerization of Maleimides and Vinyl Ether:A Structural Study”“,Maromolecules 1998,(31),pp.5681-89中找到。
贫电子交联基团包括马来酰亚胺、马来酸酯、富马酸酯和其它不饱和酯。电子供体基团包括乙烯基醚、丙烯醚和其他类似的烯基醚。像这些的混合物是有利的在于各个组分在热和光化学上是稳定的,而货架寿命优异。然而,当材料组合时,混合物具有高的光化学敏感性并对于交联仅需要相对较小的UV剂量。
根据本发明的可交联材料已用作OLED器件中的发光层。器件的测量表明,所发射的光源自交联的Lomox层。
器件的结构为ITO(150nm)/PEDOT:PSS(50nm)/PVK(30nm)/Lomox发射体材料(65nm,未漂洗)/TPBI(30nm)/LiF(1nm)/Al(70nm)。两个器件(每个4×4mm的区域4个像素)采用这种结构制作。
发射体层由以蓝:绿比率90:10用绿色发射体掺杂的蓝色宿主构成。这个最终比率用具有马来酰亚胺交联剂单元的(A,化合物43)蓝色荧光团和作为宿主材料用具有乙烯基醚交联剂单元的(C,化合物58)绿色荧光团掺杂的具有乙烯基醚交联剂单元的(B,化合物41)蓝色荧光团,以A:B:C比率50:40:10进行共混的混合物而实现。
溶液可加工的PEDOT:PSS、PVK和发射体层通过旋涂沉积于预图案化玻璃/ITO基片上。以2500rpm由20mg/ml的甲苯溶液旋涂Lomox发射体材料60秒产生厚度为65nm的质量良好的均匀膜。
在旋涂之后,发射体层以5W/cm2的功率密度在氩气氛下暴露于具有280至450nm宽发射的金属卤化物灯(Dymax BlueWave 200)15秒以交联材料。这个宽谱光源覆盖了350至380nm的激发波长范围,发现在此范围内对于这些器件中使用的Lomox材料交联过程是速度最快的。在交联之后,一个器件用甲苯旋转漂洗以去除任何未交联的材料,而一个器件保持未漂洗以进行对比。TPBI电子传输层和阴极层随后通过热蒸发沉积。
用于这些简单未优化器件的电流电压和亮度数据如图5中所示。具有交联和漂洗层的器件在7.5V的电压上具有一个转变,高于此电压时器件会以超过10cd/m2的亮度发射,而在16.5V达到804cd/m2的峰值亮度。这表明这些材料的交联产生了适合OLED器件应用的不溶性发光膜。电流和亮度对于漂洗的器件都较高,因为漂洗的层更薄。
这些器件的电致发光光谱连同交联薄膜中掺杂于蓝色宿主中的绿色材料的光致发光光谱一起示于图6中。这些光谱清楚地表明,发射是绿色的,并因此由发光层中的绿色Lomox发射体产生。漂洗后发射的光谱移位很可能是因为漂洗层更薄的光学效应。
9,9’-(四氟烷基)取代的芴发射体核芯相对于相应的9,9’-烷基芴核芯的稳定性通过将每种化合物(以上表述为“Lomox蓝”和“标准蓝”)的溶液暴露于具有150W氙光源的辐射并测量荧光强度随时间的降低而进行评估。该研究的结果(图7和8)表明,9,9’-(四氟烷基)取代的芴发射体的半衰期是相应的9,9’-烷基芴的约5倍长。因此明显的是,9,9’-(四氟烷基)取代提供了增强的稳定性。
另外的研究认为,9,9’-(四氟烷基)取代的芴发射体核芯和相应的9,9’-烷基芴核芯的吸收和发射特性是类似的,并且在芴9-位上的氟烷基链对溶液中的各个反应性荧光团的荧光效率没有不利影响。
荧光量子产率(φF)是吸收的光子与通过荧光发射的光子的比率。它反映了激发态通过荧光而不是非辐射机制失活的可能性。在一般情况下,φF越高越好,因为它会导致更有效的OLED器件。认为,短的荧光寿命对于OLED可能是更理想的,因为分子处于激发态的时间更短,而因此,分子经受不想要的非辐射衰变和荧光效率降低的机会更少。Lomox蓝化合物据证明在甲苯溶液中具有的荧光寿命为15ns,其比对于在甲苯溶液中显示出75ns的荧光寿命的标准蓝所观察显著更短,这表明氟烷基芴体系在这个方面具有另外的优势。
表1.在标准蓝和Lomox蓝的固态中的ΦF的对比
本发明的化合物在溶液中和固态下表现出高φF(使用积分球进行固态φF测量)并在通过循环伏安法研究时在DCM溶液中显示出有效的电化学稳定性和可逆性。
附图说明
图1表明根据本发明的化合物能够在性质上是液晶。化合物23在110℃下的这种偏振光显微镜图像显示出具有线状纹理的纹影并表明分子采取了向列间相。
图2在第一图和第二图中分别显示了引入本发明的发射体作为PVK宿主中的掺杂剂的器件的:J-V(左)和L-V(右)数据。
图3显示了引入Lomox发射体作为PVK宿主中的掺杂剂的器件的标准化电致发光光谱。普通PVK器件的光谱包括在内用于对比。
图4显示了引入Lomox绿发射体作为掺杂剂和Lomox蓝发射体作为宿主材料的标准化电致发光光谱。
图5显示了具有由根据本发明的交联的材料构成的发光层的器件的电流密度与电压J-V(左)和L-V(右)的数据的图。
图6显示了具有由交联的Lomox材料构成的发光层的器件的标准化电致发光光谱,以及在用于对比的交联的薄膜中Lomox蓝宿主中掺杂的Lomox绿发射体的光致发光光谱。
图7显示了在暴露于150W氙光源的光的长度变化之后Lomox蓝/化合物1(图7A)和标准蓝/化合物2(图7B)在甲苯溶液中的荧光光谱。
图8显示了Lomox蓝/化合物1(橙色)和标准蓝/化合物2(蓝色)在甲苯溶液中相对于通过150W的氙光源辐照的时间在391nm下的荧光峰值的剩余荧光强度。
合成实施例
本发明的化合物可以通过本领域普通技术人员公知的有机合成方法中的常用技术进行合成。如何能够合成这些化合物的示例性实例如下所示。正如所理解的是,这些材料的性质允许采纳模块化的合成方法并且氟烷基芴核芯能够通过标准化学技术整合到材料范围内。按照这种方式材料,例如,核芯A的所有组分、各种柔性连接基/间隔基团(S1、S1a、S2、S2a和S3)和各种交联剂和含有结构(D、B1、B2和B3)的交联剂的性质能够容易地调节以精准调整本体材料的性质如熔点、液体结晶度和发光特性。以下提供的实施例是仅以实例的方式而不以任何方式限制本发明的范围。
方案1 9,9-二氟烷基芴-2,7-联苯衍生物的合成
八氟化酮1
在氩气中-78℃下将甲基锂溴化锂复合物(6.6ml,9.9mmol,1.5mM在乙醚中)10min内滴加至碳酸二乙酯(1.0ml,8.3mmol)和1-溴-1,1,2,2-四氟辛烷(3.2ml,20.5mmol)在干乙醚(150ml)中的搅拌溶液中。反应混合物在-78℃下搅拌10min,此后在-78℃下10min内滴加将另一等分的甲基锂溴化锂复合物(6.6ml,9.9mmol,1.5mM在乙醚中)。反应混合物保持-78℃下再搅拌一个小时。随后用浓HCl(6ml)在-78℃下淬灭反应而容许混合物温热至室温。溶液用2M HCl(100ml)洗涤以及水层用乙醚(2×50ml)萃取。合并的有机物进行干燥(MgSO4),过滤并在减压下蒸掉溶剂。粗产物混合物通过真空蒸馏进行纯化。所需产物1作为无色油获得(160℃,1.3mbar,2.0g,5.0mmol,61%)。1H NMR(400MHz,CDCl3),δ=0.90(6H,t,J=7Hz,CH3),1.29-1.41(12H,m,CH2),1.54-1.62(4H,m,CH2),1.98-2.11(4H,m,CH2);13C NMR(100MHz,CDCl3),δ=14.1(CH3),20.3(t,J=3.3Hz,CH2),22.6(CH2),29.0(CH2),30.6(t,J=22.3Hz,CH2),31.5(CH2),110.6(t,J=38.5Hz,CF2),113.3(t,J=38.5Hz,CF2),184.5(m,C=O);19F NMR(376MHz,CDCl3),δ=-118.3(CF2),-113.2(CF2)。
八氟化叔醇2
在氩气中-78℃下将正丁基锂(520μL,1.30mmol,2.5M在己烷中)30min内滴加至2-碘联苯(0.18ml,1.00mmol)在干己烷(10ml)中的搅拌溶液中。混合物在-78℃下搅拌30min并随后容许温热至室温。混合物随后室温下搅拌30min之后冷却至-78℃。将八氟化酮(0.50g,1.26mmol)溶解于干己烷(10ml)中并随后在-78℃下加入到反应混合物。容许反应混合物温热至室温并搅拌16h。在清晨反应混合物用5%HCl(30ml)淬灭,分离层以及用乙醚(2×30ml)萃取水层。合并的有机物用5%HCl(30ml)和水(30ml)洗涤。它们随后进行干燥(MgSO4),过滤并在减压下蒸掉溶剂。粗产物通过快速色谱法(己烷/5%在己烷中的EtOAc)进行纯化,这获得作为无色油的产物2(0.48g,0.87mmol,87%)。1H NMR(400MHz,CDCl3),δ=0.90(6H,t,J=7Hz,CH3),1.26-1.33(12H,m,CH2),1.43-1.51(4H,m,CH2),1.90-2.03(4H,m,CH2),3.20(1H,s,OH),7.09-7.12(1H,m,Ar-H),7.33-7.36(2H,m,Ar-H),7.39-7.42(5H,m,Ar-H),7.80(1H,br s,Ar-H);19F NMR(376MHz,CDCl3),δ=-114.9(2F,d,J=286Hz,CF2),-113.8(2F,d,J=284Hz,CF2),-110.8(2F,d,J=260Hz,CF2),-108.8(2F,d,J=260Hz,CF2),MS(ASAP+):535.2(100,[M-H2O+H]+),553.3(30,[M+H]+)。
9,9-二(1,1,2,2-四氟辛烷)芴3
将八氟化叔醇(2.0g,3.62mmol)溶解于亚硫酰氯(7.9ml,109mmol)和吡啶(1.6ml,19.5mmol)的混合物中。将反应混合物在氩气中100℃下搅拌3天,此后容许反应混合物冷却至室温并滴加至冰冷水(200ml)中。随后用乙醚(3×100ml)萃取产物。将合并的有机物进行干燥(MgSO4),过滤并在减压下蒸掉溶剂。粗产物通过快速色谱法(己烷/5%在己烷中的EtOAc)进行纯化,这获得作为无色油的产物3(0.3g,0.56mmol,15%)。1H NMR(400MHz,CDCl3),δ=0.80(6H,t,J=7Hz,CH3),1.06-1.30(16H,m,CH2),1.46-1.58(4H,m,CH2),7.33(2H,td,J=7.7Hz,1.2Hz,Ar-H),7.50(2H,td,J=7.5Hz,1.0Hz,Ar-H),7.75(2H,br d,J=7.5Hz,Ar-H),7.79(2H,br d,J=7.8Hz,Ar-H);19F NMR(376MHz,CDCl3),-109.7(CF2),-107.9(CF2)。
2,7-二溴-9,9-二(1,1,2,2-四氟辛烷)芴4
将N-溴琥珀酰亚胺(0.23g,1.31mmol)加到9,9-二(1,1,2,2-四氟辛基)芴(0.35g,0.66mmol)在12ml 5:1乙酸浓硫酸溶液中的搅拌溶液中。反应混合物在65℃下搅拌16h。在冷却至室温之后,将反应混合物倾倒入10%NaOH溶液(100ml)中并将产物用乙醚(3×50ml)从水溶液中萃取出来。合并的有机物用10%NaOH溶液(50ml)和水(50ml)洗涤。有机层随后进行干燥(MgSO4),过滤并在减压下除去溶剂。粗产物混合物通过快速色谱法(己烷)进行纯化。这获得作为白色晶体固体的产物4(0.22g,0.32mmol,48%)。1H NMR(400MHz,CDCl3),δ=0.83(6H,t,J=7.0Hz,CH3),1.13-1.26(16H,m,CH2),1.62-1.76(4H,m,CH2),7.57(2H,d,J=8.2Hz,Ar-H),7.62(2H,dd,J=8.2,1.7Hz,Ar-H),7.88(2H,br d,J=1.7Hz,Ar-H);19F NMR(376MHz,CDCl3),δ=-109.1(4F,t,J=19Hz,CF2),-107.2(4F,s,CF2)。
4’-辛氧基联苯-4-硼酸5
将碳酸钾(9.43g,68.3mmol)分批加入到4-溴-4’-羟基联苯(10.0g,40.1mmol)和1-溴辛烷(9.0ml,52.2mmol)在丁酮(100ml)中的搅拌溶液中。反应混合物回流搅拌16h。在清晨容许反应混合物冷却至室温并通过过滤除去白色固体。母液减压下蒸发至干燥而粗产物通过乙醇重结晶进行纯化。这获得作为白色固体的产物(11.6g,32.2mmol,80%)。将4-溴-4’-辛氧基联苯(4.0g,11.1mmol)溶解于干THF(100ml)中并冷却至-78℃。将正丁基锂(5.3ml,13.3mmol,2.5M在己烷中)滴加到该溶液中以及将反应混合物在氩气中-78℃下搅拌1h。随后-78℃下滴加三甲基硼酸酯(2.47ml,22.1mmol)。随后容许反应混合物温热至室温并搅拌18h。在清晨加入2M HCl溶液(10ml)以及将混合物搅拌1h。产物用乙醚(2×30ml)萃取以及将合并的有机物进行干燥(MgSO4),过滤并在减压下除去溶剂。粗产物在己烷下研磨以获得作为白色固体的产物5(1.88g,5.8mmol,52%)。1H NMR(400MHz,SO(CD3)2),δ=0.86(3H,t,J=7.0Hz,CH3),1.26-1.45(10H,m,CH2),1.72(2H,五重峰,J=6.6Hz,CH2),3.99(2H,t,J=6.5Hz,CH2),7.00(2H,dt,J=8.9,2.1Hz,Ar-H),7.57-7.62(4H,m,Ar-H),7.84(2H,d,J=8.3Hz,Ar-H),8.03(2H,br s,OH)。
化合物6
将四(三苯基膦)钯(37mg,10mol%)加到2,7-二溴-9,9-二(四氟辛基)芴(0.22g,0.32mmol)和4’-辛氧基联苯-4-硼酸(0.26g,0.79mmol)在脱气的1,2-二甲氧基乙烷(15ml)和脱气的20%Na2CO3水(8ml)溶液中的搅拌溶液中。将反应混合物在氩气中回流搅拌18h。完成之后,容许混合物冷却至室温并减压下除去1,2-二甲氧基乙烷。含水层用水(100ml)稀释并随后用二氯甲烷(100ml,2×50ml)萃取。合并的有机物用盐水(70ml)洗涤,干燥(MgSO4),过滤并在减压下蒸发至干燥。粗产物混合物通过快速色谱法(20%在己烷中的二氯甲烷)进行纯化并随后用二氯甲烷和乙醇重结晶以获得作为白色微晶固体的产物6(0.25g,0.23mmol,72%)。1H NMR(400MHz,CDCl3),δ=0.78(3H,t,J=7.0Hz,CH3),0.90(3H,t,J=7.0Hz,CH3),1.06-1.53(36H,m,CH2),1.56-1.65(4H,m,CH2),1.82(4H,五重峰,J=6.8Hz,CH2),4.02(4H,t,J=6.5Hz,CH2),7.01(4H,dt,J=8.8,2.1Hz,Ar-H),7.58(4H,dt,J=8.8,2.1Hz,Ar-H),7.67(4H,d,J=8.6Hz,Ar-H),7.72(4H,d,8.6Hz,Ar-H),7.79(2H,dd,J=8.0,1.6Hz,Ar-H),7.84(2H,d,J=8Hz,Ar-H),8.08(2H,br s,Ar-H);13C NMR(100MHz,CDCl3),δ=14.1(CH3),14.3(CH3),20.3,22.5(CH2),22.8(CH2),26.2(CH2),28.8(CH2),29.4(CH2),29.5(2x CH2),31.4(CH2),32.0(CH2),68.3(CH2),115.0,120.3,127.3,127.6,128.2,128.7,133.1,139.1,140.1,140.3,141.1,159.0;19F NMR(376MHz,CDCl3),δ=-109.4(CF2),-107.7(CF2);HRMS(ASAP+):m/z理论值[M+H]+=1095.6266,实测值:1095.6240。
方案2 9,9-二氟烷基芴-2,7-苯并[c]-1,2,5-噻二唑衍生物的合成
4-溴-7-(4-辛氧基苯基)-2,1,3-苯并噻二唑8
将4-辛氧基苯基硼酸(1.02g,0.0041mol)、4,7-二溴苯并[c]-1,2,5-噻二唑(化合物7,1.20g,0.0041mol)、K2CO3(1.12g,0.0082mol)、甲苯(30ml)和水(15ml)都加入到3-口圆底烧瓶中并以真空泵辅助,将系统排空,并用氮气填充3次。随后,加入Pd(PPh3)4(0.24g,0.20mmol)并将反应混合物加热至90℃过夜。将反应混合物倾倒入分液漏斗中,向其中加入水(10ml)并另加入甲苯(10ml)。有机层减压浓缩,随后使用甲苯共沸干燥。粗产物通过重力柱层析(硅胶)使用梯度洗脱(30%在己烷中的CH2Cl2/50%在己烷中的CH2Cl2)进行纯化而获得作为黄色粉末的8(0.65g,38%)。1H NMR(400MHz,CDCl3):δ(ppm)0.89(t,3H),1.26-1.52(m,10H),1.82(五重峰,2H),4.04(t,2H),7.05(d,2H),7.53(d,1H),7.85(d,2H),7.90(d,1H)。
4-(4-(辛氧基)苯基)-7-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊环-2-基)-2,1,3-苯并噻二唑9
将化合物8(0.60g,0.0014mol)、双戊酰二硼(0.40g,0.0016mol)、乙酸钾(0.42g,0.0043mol)和PdCl2(dppf)(0.06g,0.07mmol)加入到圆底烧瓶中并用氮气吹扫。脱气的干二氧六环(15ml)通过注射器加入并将反应混合物加热至80℃持续14h。将反应混合物进行过滤并将二氧六环使用旋转蒸发器除去。粗产物通过重力柱层析(硅胶)使用梯度洗脱(CH2Cl2和5%CH2Cl2中的乙酸乙酯)进行纯化以获得作为黄褐色固体的9(0.34g,51%)。1H NMR(400MHz,CDCl3):δ(ppm)0.92(t,3H),1.28-1.54(m,10H),1.48(s,12H),1.85(五重峰,2H),4.06(t,2H),7.08(d,2H),7.68(d,1H),7.94(d,2H),8.26(d,1H)。
化合物10
将四(三苯基膦)钯(10mg,8.7μmol)加到2,7-二溴-9,9-(1’1’2’2’-四氟辛基)芴(61mg,87.7μmol)和化合物9(90mg,0.19mmol)在脱气的1,2-二甲氧基乙烷(15ml)和脱气的20%Na2CO3溶液(8ml)的搅拌溶液中。将反应混合物在氩气中搅拌回流过夜。完成之后,容许混合物冷却至至室温以及在减压下除去1,2-二甲氧基乙烷。随后用二氯甲烷(100ml,2×50ml)萃取含水层并将合并的有机物用盐水(70ml)洗涤,干燥(MgSO4),过滤并减压蒸发。粗产物混合物通过快速色谱法(30%在己烷中的二氯甲烷)并随后用二氯甲烷乙醇混合物重结晶进行纯化。这获得作为黄色结晶固体的产物10(69mg,57.0μmol)m.p.120℃。1H NMR(400MHz,CDCl3):δ(ppm)0.75(t,J=7.0Hz,6H),0.91(t,J=6.9Hz,6H),1.06-1.16(m,12H),1.20-1.27(m,4H),1.29-1.41(m,16H),1.51(quin,J=7.2Hz,4H),1.58-1.69(m,4H),1.85(五重峰,J=7.0Hz,4H),4.07(t,J=6.6Hz,4H),7.10(td,J=8.8,2.5Hz,4H),7.79(d,J=7.4Hz,2H),7.87(d,J=7.4Hz,2H),7.95(td,J=8.8,2.4Hz,4H),7.98(d,J=8.0Hz,2H),8.28(dd,J=8.0,1.5Hz,2H)8.40(br s,2H);质量(MALDI)=1210.5(M+)。
方案3 9,9-二氟烷基芴-2,7-噻吩/苯并[c]-1,2,5-噻二唑衍生物的合成
4,7-二-2-噻吩基-2,1,3-苯并噻二唑12
将具有4,7-二溴苯并[c]-1,2,5-噻二唑(化合物11、2.00g,0.0068mol)、2-(三丁基甲锡烷基)噻吩(5.59g,0.0150mol)和甲苯(30ml)的反应混合物的溶液用氮鼓泡通过以对溶剂脱氧。在氮气氛下加入Pd(PPh3)4(0.50g,0.43mmol)并在90℃下加热搅拌3天。将甲苯减压除去以及粗产物通过重力柱层析(硅胶)使用洗脱剂30%在己烷中的CH2Cl2接着2×己烷重结晶进行纯化以获得作为亮红色晶体的12(1.21g,59%)。1H NMR(400MHz,CDCl3):δ(ppm)7.20(dd,2H),7.45(dd,2H),7.88(s,2H),8.12(dd,2H)。
4-(5-溴-2-噻吩基)-7-(2-噻吩基)-2,1,3-苯并噻二唑13
在冰水浴中搅拌下将N-溴琥珀酰亚胺(由水重结晶,0.59g,3.3mmol)缓慢加入化合物12(1.00g,3.3mmol)在干DCM(40ml)和乙酸(20ml)中的溶液中。所得的反应混合物室温搅拌过夜。将反应混合物加入到DCM(500ml)中并加入饱和NaHCO3水溶液(100ml)。有机层用水(2×200ml)洗涤,干燥(MgSO4),过滤并减压浓缩。粗产物通过重力柱层析(硅胶)使用20%在己烷中的CH2Cl2进行纯化以产生作为红色粉末的13(0.75g,60%)。1H NMR(400MHz,CDCl3):δ(ppm)7.16(d,1H),7.22(dd,1H),7.47(dd,1H),7.79-7.82(m,2H),7.87(d,1H),8.13(dd,1H)。
4-(5-(4-辛氧基苯基-2-噻吩基)-7-(2-噻吩基)-2,1,3-苯并噻二唑14
将4-辛氧基苯基硼酸(0.69g,2.77mmol)、化合物13(0.70g,1.85mmol)、K2CO3(1.27g,9.23mmol)、甲苯(40ml)、乙醇(5ml)和水(20ml)都加入到3-口圆底烧瓶中并以真空泵辅助将系统排空,并用氮气填充3次。随后,加入Pd(PPh3)4(0.10g,0.09mmol)并将反应混合物加热至90℃过夜。将反应混合物倾倒入分液漏斗中,其中加入水(10ml)以及另外的甲苯(10ml)。有机层减压浓缩而随后共沸干燥,粗产物通过快速柱层析(硅胶)使用30%在己烷中的CH2Cl2作为洗脱剂进行纯化以获得作为红色粉末的14(0.55g,59%)。1H NMR(400MHz,CDCl3):δ(ppm)0.92(t,3H),1.28-1.40(m,8H),1.48(五重峰,2H),1.83(五重峰,2H),4.03(t,2H),6.97(d,2H),7.24(dd,1H),7.33(d,1H),7.48(dd,1H),7.65(d,2H),7.90(d,2H),8.12(d,1H),8.15(dd,1H)。
4-(5-(4-辛氧基苯基-2-噻吩基)-7-(5-(三丁基甲锡烷基)-2-噻吩基)-2,1,3-苯并噻二唑15
所有玻璃器皿使用前置于热烘箱中过夜并在氮气氛下冷却至室温。将正丁基锂(2.5M在己烷中,0.17ml,0.41mmol)在-78℃下慢慢地滴加到2,2,6,6-四甲基哌啶(0.07g,0.50mmol)在干THF(5ml)中的溶液中并搅拌15min。将反应混合物通过除去干冰/丙酮浴而温热至室温并在室温下搅拌15min,并再次回冷至-78℃。将化合物14(0.20g,0.40mmol)在干THF(15ml)中的溶液滴加至锂盐中并在-78℃下搅拌1min。最后,反应用三丁基氯化锡(0.15g,0.13ml,0.46mmol)淬灭并室温搅拌过夜。加入水并使用二氯甲烷进行后处理,干燥(K2CO3),过滤并减压浓缩。粗产物15无需进一步纯化或表征直接用于下一步骤。
化合物16
将四(三苯基膦)钯(5mg,10mol%)加到2,7-二溴-9,9-二(1’1’2’2’-四氟辛基)芴(35mg,50μmol)和化合物15(100mg,0.13mmol)在脱气的甲苯(5ml)中的搅拌溶液中。将反应混合物搅拌回流过夜。在清晨容许反应混合物冷却至室温并随后用水(20ml)洗涤以及水层用二氯甲烷(2×20ml)洗涤。合并的有机物进行干燥(MgSO4),过滤并减压下除去溶剂。粗产物通过快速色谱法(5%至40%在己烷中的二氯甲烷)进行纯化,这获得作为暗红色固体的产物16(34mg,44%)。1H NMR(400MHz,CDCl3):δ(ppm)0.78(t,J=7.0Hz,6H),0.90(t,J=6.9Hz,6H),1.09-1.40(m,32H),1.44-1.52(m,4H),1.62-1.75(m,4H),1.81(五重峰,J=7.0Hz,4H),4.00(t,J=6.6Hz,4H),6.95(td,J=8.8,2.5Hz,4H),7.31(d,J=3.9Hz,2H),7.52(d,J=3.9Hz,2H),7.63(td,J=8.8,2.5Hz,4H),7.77(d,J=8.1Hz,2H),7.84-7.93(m,6H),8.11-8.16(m,6H);液晶转变(℃):[Cr玻璃N 170 I]。质量(MALDI)=1538.4(M+)。
方案4 具有富电子交联剂的氟烷基芴衍生物23的合成
2,7-二(4-辛氧基联苯-4’-基)-9-9-二(1,1,2,2-四氟戊烷)芴19
将4-辛氧基联苯-4’-基硼酸(化合物18,0.52g,1.59mmol)、2,7-二溴-9,9-二(1,1,2,2-四氟戊烷)芴(化合物17,0.42g,0.69mmol)、K2CO3(0.48g,3.45mmol)、DME(15ml)和水(5ml)全部加入到3-口圆底烧瓶中并以真空泵辅助将系统排空,并用氮气填充3次。随后,加入Pd(PPh3)4(0.08g,0.07mmol)以及将反应混合物回流下搅拌过夜。将反应混合物倾倒入分液漏斗中,向其中加入水(100ml)和CH2Cl2(100ml),水层用CH2Cl2(50ml)洗涤以及合并的有机层用水(100ml)洗涤并干燥(MgSO4)。在滤掉MgSO4之后粗产物通过重力柱层析(硅胶)使用梯度洗脱(20%在己烷中的CH2Cl2/50%在己烷中的CH2Cl2)进行纯化以获得白色粉末的19(0.45g,64%)。液晶转变(℃):[Cr 151(N 144)I]。1H NMR(400MHz,CDCl3):δ(ppm)0.77(三重峰,6H),0.92(三重峰,6H),1.24-1.55(m,24H),1.64(五重峰,4H),1.85(五重峰,4H),4.04(三重峰,4H),7.03(d,4H),7.61(d,4H),7.68-7.87(m,12H),8.10(s,2H).液晶转变(℃):[Cr 151(N 144)I]。
2,7-二(4-羟基联苯-4’-基)-9-9-二(1,1,2,2-四氟戊烷)芴20
在冰/盐/水浴中将三溴化硼(0.50g,0.19ml,1.98mmol)缓慢加入化合物19(0.40g,0.40mmol)在干CH2Cl2(10ml)中的溶液中。将反应混合物温热至室温并搅拌过夜。在TLC指示完全去烷基化之后将反应混合物倾倒入冰水混合物(50ml)中并搅拌另一个小时。含水层用CH2Cl2(2×50ml)洗涤以及合并的有机萃取物用水洗涤,干燥(MgSO4)并过滤。在减压浓缩之后获得作为灰白色粉末的20(0.25g,80.6%)。1H NMR(400MHz,CDCl3):δ(ppm)0.77(t,6H),1.29(五重峰,8H),4.85(s,2H),6.97(d,4H),7.57(d,4H),7.67-7.87(m,12H),8.10(s,2H)。
8-溴-1-乙烯氧基辛烷22
使用热风枪在真空下干燥配备有水冷凝器的单口圆底烧瓶。在冷却至室温之后加入8-溴-1-辛醇(化合物21)、醋酸乙烯酯(干燥并蒸馏的)、Na2CO3(80℃下真空干燥5h)和干甲苯(20ml)并用氮气吹扫。加入催化剂[Ir(cod)Cl]2并在100℃下搅拌反应混合物2h。将反应混合物冷却至50℃并在高真空(油泵)下除去甲苯以及粗产物通过短硅胶填充柱使用20%在己烷中的DCM作为溶剂纯化以获得黄色油(1.50g,60%)。1H NMR(400MHz,CDCl3):δ(ppm)1.30-1.45(m,8H),1.65(五重峰,2H),1.85(五重峰,2H),3.40(t,2H),3.67(t,2H),3.97(dd,1H),4.16(dd,1H),6.46(dd,1H)。
化合物23
在氮气氛下将化合物20(0.25g,0.32mmol)、化合物22(0.19g,0.79mmol)、Cs2CO3(0.52g,1.59mol)和四丁基溴化铵(小勺)在丁酮(10ml)中搅拌回流过夜。过滤掉盐,减压下除去丁酮以及将粗产物静置一周时间而结晶析出。将所得的固体用乙醇洗涤并过滤以获得作为白色粉末的23(0.18g,51.4%)。液晶转变(℃):[Cr 85 N 121 I]。1H NMR(400MHz,CDCl3):δ(ppm)0.77(三重峰,6H),1.30(五重峰,8H),1.35-1.55(m,16H),1.69(五重峰,4H),1.85(五重峰,4H),3.71(三重峰,4H),4.00(dd,2H),4.04(三重峰,4H),4.20(dd,2H),6.50(dd,2H),7.03(d,4H),7.61(d,4H),7.68-7.87(m,12H),8.10(s,2H)。
方案5 与马来酰亚胺(贫电子)交联剂可交联的芴衍生物的合成
3,6-内,外-氧-Δ4-四氢酞二酰亚胺25
将马来酰亚胺(24,2.50g,0.0258mol)和呋喃(10ml)在乙酸乙酯(20ml)中的溶液室温搅拌3天。所得的沉淀经过过滤并真空干燥以获得白色粉末(3.00g,71%)。产物25作为内-和外-加成物(1.6:1)的混合物而获得。1H NMR(400MHz,CDCl3):δ(ppm)内产物:3.6(s,2H),5.35(s,2H),6.6(s,2H),外产物:2.9(s,2H),5.35(s,2H),6.6(s,2H)。
8-(3,6-内-氧-Δ4-四氢酞二酰亚胺)溴辛烷26
将化合物25(0.76g,0.0046mol)、1,8-二溴辛烷(5.00g,0.0184mol)、K2CO3(3.18g,0.0230mol)和DMF(10ml)在55℃氮气氛下搅拌过夜。减压除去DMF并加入DCM以及将盐过滤掉。粗产物通过柱层析(硅胶,30%在己烷中的乙酸乙酯/50%在己烷中的乙酸乙酯)进行纯化以获得作为结晶过夜的粘性液体的26(0.40g,24%)。1H NMR(400MHz,CDCl3):δ(ppm)外产物:1.20-1.60(m,10H),1.85(五重峰,2H),2.9(s,2H),3.40(三重峰,2H),3.50(三重峰,2H),5.35(s,2H),6.6(s,2H)。
化合物28
将化合物26(0.30g,0.83mmol)、化合物27(0.20g,0.28mmol)、TBAB(小勺(spatula tip))和Cs2CO3(0.45g,1.38mmol)在丁酮(20ml)中室温搅拌长3天并随后在氮气氛下50℃搅拌5h。过滤掉盐,减压下除去丁酮以及将粗产物进行柱层析(硅胶,50%在己烷中的乙酸乙酯/100%乙酸乙酯)以获得作为粘性液体的28(0.25g,71%)。1H NMR(400MHz,CDCl3):δ(ppm)外产物:0.70-0.85(m,10H),1.10-1.60(m,40H),1.85(五重峰,4H),2.10(m,4H),2.85(s,4H),3.50(三重峰,4H),4.00(三重峰,4H),5.35(s,4H),6.6(s,4H),7.00(d,4H),7.60-7.90(m,18H)。
化合物29
将化合物5(0.30g,0.23mmol)在甲苯(10ml)中回流过夜至完全消除呋喃。将甲苯减压除去以获得缓慢结晶的粘性液体(0.27g,100%)。1H NMR(400MHz,CDCl3):δ(ppm)0.71-0.84(m,10H),1.04-1.65(m,40H),1.83(五重峰,4H),2.08(m,4H),3.55(三重峰,4H),4.04(三重峰,4H),6.71(s,4H),7.02(d,4H),7.59-7.82(m,18H)。液晶转变(℃):[Cr 74 N 102 I]。质量(MALDI)=1140.6(M+)。
方案6 与马来酸酯(贫电子)交联剂可交联的芴衍生物33的合成
化合物32
将2-(8-溴辛氧基)四氢-2H-吡喃(化合物30,0.97g,3.30mmol)、化合物27(0.80g,1.10mol)和K2CO3(0.76g,5.50mol)的反应混合物在干DMF(10ml)中100℃下搅拌过夜。将反应冷却至室温并将DMF减压除去。将粗产物溶解于DCM中并过滤掉盐。在除去DCM之后将粗产物(化合物31)溶解于甲醇(20ml)中并用浓硫酸(1ml)回流。在一个小时之后将反应混合物冷却至室温并将生成的沉淀过滤并用大量的水洗涤。在真空干燥之后获得所需产物32是白色粉末(0.80g,74%,两个步骤内)。1H NMR(400MHz,CDCl3):δ(ppm)0.71-0.84(m,10H),1.06-1.74(m,40H),1.85(五重峰,4H),2.08(m,4H),3.69(三重峰,4H),4.05(三重峰,4H),7.02(d,4H),7.60-7.82(m,18H),-OH质子未检测。
化合物33
将DCC(0.24g,1.16mmol)在氮气氛下分批加入到化合物32(0.20g,0.20mmol)、马来酸单乙酯(0.37g,2.8mmol)和DMAP(0.10g,0.82mmol)在干CH2Cl2(6ml)中的冷(0℃)溶液中。将反应混合物搅拌20h并将生成的DCU过滤并减压除去CH2Cl2。获得的残余物通过柱层析(硅胶)梯度洗脱(10%在己烷中的乙酸乙酯/20%在己烷中的乙酸乙酯)进行纯化以获得粘性油,其缓慢结晶成白色低熔点固体(0.10g,40%)。1H NMR(400MHz,CDCl3):δ(ppm)0.70-0.83(m,10H),1.05-1.56(m,42H),1.72(五重峰,4H),1.85(五重峰,4H),2.08(m,4H),4.04(t,4H),4.22(t,4H),4.28(五重峰,4H),6.26(s,4H)7.02(d,4H),7.60-7.82(m,18H)。液晶转变(℃):[Cr 43 N 78 I]。质量(MALDI)=1234.7(M+)。
方案7 与富马酸酯(贫电子)交联剂可交联的芴衍生物34的合成
化合物34
将DCC(0.24g,1.16mmol)在氮气氛下分批加入化合物32(0.20g,0.20mmol)、富马酸单甲酯(0.26g,2.03mmol)和DMAP(0.10g,0.82mmol)在干DCM(6ml)中的冷(0℃)溶液中。将反应混合物搅拌过夜并将生成的DCU过滤并减压除去DCM。获得的残余物通过柱层析梯度洗脱(硅胶,10%在己烷中的乙酸乙酯/20%在己烷中的乙酸乙酯)进行纯化以获得几天内缓慢结晶的粘性油。随后产物用己烷碾磨并过滤以获得作为白色粉末的34(0.15g,60%)。1H NMR(400MHz,CDCl3):δ(ppm)0.72-0.84(m,10H),1.06-1.56(m,36H),1.72(五重峰,4H),1.85(五重峰,4H),2.08(m,4H),3.84(s,6H),4.05(t,4H),4.24(t,4H),6.89(s,4H)7.03(d,4H),7.59-7.83(m,18H)。质量(MALDI)=1206.7(M+)。液晶转变(℃):[Cr 76 N 107 I]。
方案7 与乙烯基醚(富电子)交联剂可交联的芴衍生物35的合成
化合物35
在氮气氛下将化合物27(0.21g,0.29mmol)、2-(8-溴辛氧基)乙烯基醚(0.16g,0.66mmol)和K2CO3(0.20g,1.44mmol)在DMF(10ml)中90℃下搅拌过夜。过滤掉盐,减压除去DMF并将粗产物进行柱层析(硅胶,20%在己烷中的DCM)以获得作为白色粉末的35(0.05g,16.6%)。1H NMR(400MHz,CDCl3):δ(ppm)0.74-0.84(m,10H),1.07-1.56(m,36H),1.71(五重峰,4H),1.85(五重峰,4H),2.08(m,4H),3.72(t,4H),4.00(dd,2H),4.04(t,4H),4.20(dd,2H),6.50(dd,2H),7.02(d,4H),7.60-7.82(m,18H)。质量(MALDI)=1034.7(M+);液晶转变(℃):[Cr 82 N 155 I]。
与乙烯基醚和马来酰亚胺交联基团可交联的氟烷基芴衍生物的合成
化合物36
C8-二醇(化合物36)的合成;将三溴化硼(1.3ml,13.5mmol)在0℃下滴加至C8-8(化合物6,2.94g,2.68mmol)在干二氯甲烷(50ml)中的搅拌溶液中。容许反应混合物温热至室温并随后在氩气中搅拌16h。随后将冰水(50ml)加入到反应混合物并将其搅拌另一个小时,此后分离层并将水层用二氯甲烷(2×100ml)萃取。将合并的有机物进行干燥(MgSO4),过滤并减压下除去溶剂。残余物通过己烷下研磨进行纯化,这获得作为白色固体的产物36(2.01g,2.31mmol,86%)。1H NMR(400MHz,CDCl3),δ=0.78(6H,t,J=7.0Hz,CH3),1.06-1.24(14H,m,CH2),1.52-1.65(4H,m,CH2),4.77(2H,s,OH),6.95(4H,dt,J=8.7,2.1Hz,Ar-H),7.55(4H,dt,J=8.6,2.1Hz,Ar-H),7.66(4H,d,J=8.4Hz,Ar-H),7.73(4H,d,J=8.4Hz,Ar-H),7.80(2H,dd,J=8.0,1.5Hz,Ar-H),7.84(2H,d,J=8.0Hz,Ar-H),8.07(2H,br s,Ar-H);19F NMR(376MHz,CDCl3),δ=-109.0(CF2),-107.2(CF2)。
化合物37
C8-单辛烷(化合物37)的合成;将K2CO3(0.64g,4.60mmol)分批加入到C8-二醇(2.00g,2.30mmol)和1-溴辛烷(0.44g,2.30mmol)在2-丁酮(100ml)中的搅拌溶液中。将反应混合物搅拌回流18h。随后容许反应混合物冷却至室温并将剩余的K2CO3和盐过滤掉并用二氯甲烷(50ml)洗涤。滤液减压下蒸发至干燥而粗产物混合物通过快速色谱法(15%至30%在己烷中的乙酸乙酯)进行纯化。这获得作为白色固体的产物37(0.86g,0.87mmol,38%)和作为灰白色固体的起始材料36(1.09g,1.25mmol,54%)。反应使用回收的起始原料36再次进行,这会使37的总产量达1.41g,1.43mmol,62%。1H NMR(400MHz,CDCl3),δ=0.79(6H,t,J=7.0Hz,CH3),0.90(3H,t,J=7.0Hz,CH3),1.05-1.66(30H,m,CH2),1.83(2H,五重峰,J=6.8Hz,CH2),4.02(2H,t,J=6.6Hz,CH2),4.84(1H,s,OH),6.95(2H,dt,J=8.6,2.1Hz,Ar-H),7.01(2H,dt,J=8.8,2.1Hz,Ar-H),7.56(2H,dt,J=8.6,2.1Hz,Ar-H),7.59(2H,dt,J=8.8Hz,2.1Hz,Ar-H),7.65-7.68(4H,m,Ar-H),7.72-7.74(4H,m,Ar-H),7.78-7.81(2H,m,Ar-H),7.84(2H,d,J=8.0Hz,Ar-H),8.08(2H,br s,Ar-H);19F NMR(376MHz,CDCl3),δ=-108.9(CF2),-107.2(CF2)。
化合物38
二乙基-2,5-二(溴己基)氧对苯二甲酸酯(化合物38)的合成;将K2CO3(3.26g,23.6mmol)分批加入到二乙基-2,5-二羟基对苯二甲酸酯(1.00g,3.93mmol)和1,6-二溴己烷(6.1ml,39.3mmol)在2-丁酮(20ml)中的搅拌溶液中。将反应混合物回流搅拌16h,此后过滤掉K2CO3和盐并用二氯甲烷(20ml)洗涤。将滤液随后减压下蒸发至干燥并将粗产物通过快速色谱法(10%在己烷中的乙酸乙酯)进行纯化。这获得作为白色固体的产物38(1.91g,3.29mmol,84%)。1H NMR(400MHz,CDCl3),δ=1.39(6H,t,J=7.1Hz,CH3),1.50-1.52(8H,m,CH2),1.79-1.93(8H,m,CH2),3.42(4H,t,J=6.8Hz,CH2),4.01(4H,t,J=6.4Hz,CH2),4.37(4H,q,J=7.1Hz,CH2),7.34(2H,s,Ar-H)。
化合物39
G8-8-二乙酯(化合物39)的合成;将Cs2CO3(1.26g,3.88mmol)分批加入到二乙基-2,5-二(溴己基)氧对苯二甲酸酯38(0.38g,0.65mmol)和C8-单辛烷37(1.40g,1.42mmol)在干DMF(25ml)中的搅拌溶液中。将反应混合物在90℃下在氩气中搅拌18h,此后容许反应混合物冷却至室温。随后过滤掉Cs2CO3和盐并用二氯甲烷(50ml)洗涤。将滤液减压蒸发以及将残余物通过快速色谱法(二氯甲烷)纯化。这获得作为白色固体的产物39(1.15g,0.49mmol,75%)。1H NMR(400MHz,CDCl3),δ=0.78(12H,t,J=7.0Hz,CH3),0.90(6H,t,J=6.9Hz,CH3),1.06-1.66(74H,m,CH2+CH3),1.79-1.90(12H,m,CH2),4.00-4.06(12H,m,CH2),4.38(4H,q,J=7.1Hz,CH2),6.99-7.01(8H,m,Ar-H),7.36(2H,s,Ar-H),7.57-7.59(8H,m,Ar-H),7.66-7.68(8H,m,Ar-H),7.71-7.73(8H,m,Ar-H),7.78-7.84(8H,m,Ar-H),8.07(4H,br s,Ar-H);19F NMR(376MHz,CDCl3),δ=-108.84(CF2),-107.21(CF2);MS(MALDI+):m/z理论值[M+H]+=2384.2,实测值=2384.5。
化合物40
G8-8-二酸(化合物40)的合成;将1M NaOH水溶液(10ml)加到G8-8-二乙酯39(1.15g,0.49mmol)在四氢呋喃(12ml)中的搅拌溶液中。将反应混合物在50℃下搅拌3天。一旦完成,将溶液用2M HCl酸化并随后用二氯甲烷(3×50ml)萃取。将合并的有机物进行干燥(MgSO4),过滤并减压下除去溶剂以获得作为灰白色固体的所需产物40(1.04g,0.45mmol,91%)。1H NMR(400MHz,CDCl3),δ=0.78(12H,t,J=7.0Hz,CH3),0.90(6H,t,J=6.9Hz,CH3),1.06-1.66(68H,m,CH2),1.79-2.02(12H,m,CH2),4.00-4.06(8H,m,CH2),4.33(4H,t,J=6.6Hz,CH2),6.98-7.01(8H,m,Ar-H),7.57-7.59(8H,m,Ar-H),7.66-7.73(16H,m,Ar-H),7.78-7.84(8H,m,Ar-H),7.89(2H,s,Ar-H),8.08(4H,br s,Ar-H),11.09(2H,br s,OH);19F NMR(376MHz,CDCl3),δ=-108.93(CF2),-107.20(CF2)。
化合物41
G8-8-VE(化合物41)的合成;将N,N’-二环己基碳二酰亚胺(0.16g,0.76mmol)在0℃下加到G8-8-二酸40(0.45g,0.19mmol)和4-二甲基氨基吡啶(9.3mg,0.076mmol)在干二氯甲烷(7ml)中的搅拌溶液中。随后将混合物在0℃下在氩气中搅拌1h,此后加入在干二氯甲烷(3ml)中的1,4-丁二醇乙烯基醚(0.089g,0.76mmol)并容许反应混合物温热至室温并置于氩气中搅拌18h。此后溶液用二氯甲烷(60ml)稀释并随后用饱和NaHCO3(2×50ml)和水(50ml)洗涤。随后将有机层进行干燥(MgSO4),过滤并减压下除去溶剂。粗产物通过快速色谱法(二氯甲烷)进行纯化。这获得作为白色固体的所需产物41(0.25g,0.098mmol,51%)。1H NMR(400MHz,CDCl3),δ=0.78(12H,t,J=7.0Hz,CH3),0.90(6H,t,J=6.9Hz,CH3),1.06-1.65(68H,m,CH2),1.79-1.92(20H,m,CH2),3.74(3H,t,J=6.0Hz,CH2),3.98-4.05(14H,m,CH2+=CH2),4.18(2H,dd,J=14.4,2.0Hz,=CH2),4.36(4H,t,J=6.2Hz,CH2),6.47(2H,dd,J=14.4,6.8Hz,=CH),6.99-7.01(8H,m,Ar-H),7.37(2H,s,Ar-H),7.57-7.59(8H,m,Ar-H),7.65-7.73(16H,m,Ar-H),7.78-7.84(8H,m,Ar-H),8.07(4H,br s,Ar-H);19F NMR(376MHz,CDCl3),δ=-108.93(CF2),-107.21(CF2);MS(MALDI+):m/z理论值[M+H]+=2524.3,实测值=2524.8。
化合物42
N-癸醇-马来酰亚胺(化合物42)的合成;将10-溴-1-癸醇(2.9g,12.4mmol)在干DMF(10ml)中的溶液在氩气下加入到呋喃保护的马来酰亚胺(2.1g,12.4mmol,作为内外异构体的混合物)和K2CO3(1.7g,12.4mmol)在干DMF(40ml)中的搅拌悬浮液中。随后将反应混合物在50℃下在氩气中搅拌16h(反应颜色转变成暗红色过夜)。此后将反应混合物倾入水(100ml)中并用乙酸乙酯(3×50ml)萃取。合并的有机物用水(100ml)和盐水(2×50ml)洗涤。随后将有机层进行干燥(MgSO4),过滤并减压下除去溶剂。油样残余物在己烷下用超声辅助进行研磨,其随后倾析。这个过程重复两次以除去任何未反应的溴-癸醇。随后将油样残余物溶解于乙醚中并过滤。滤液减压下蒸发至干燥以获得作为无色油的产物,其在室温下静置过夜进行固化(2.3g,7.2mmol,58%)。
将内/外N-癸醇-呋喃保护的马来酰亚胺(1.2g,3.6mmol)的混合物在甲苯(20ml)中加热至回流并回流搅拌18h。此后减压下除去溶剂并将粗产物通过快速色谱法(30%-50%在己烷中的乙酸乙酯)进行纯化。这获得作为白色固体的产物42(0.52g,2.1mmol,58%)。1H NMR(400MHz,CDCl3),δ=1.22-1.35(12H,m,CH2),1.54-1.61(4H,m,CH2),3.48-3.52(2H,m,CH2),3.63(2H,t,J=6.6Hz,CH2),6.68(2H,s,=CH)。
化合物43
G8-8-MI(化合物43)的合成;将N,N’-二环己基碳二酰亚胺(0.13g,0.64mmol)在0℃下加到G8-8-二酸40(0.60g,0.26mmol)和4-二甲基氨基吡啶(3mg,0.026mmol)在干二氯甲烷(7ml)中的搅拌溶液中。随后将混合物在氩气下载0℃下搅拌1h,此后加入在干二氯甲烷(3ml)中的N-癸醇-马来酰亚胺42(0.26g,1.0mmol)并容许反应混合物温热至室温并置于氩气中搅拌18h。随后将溶液用二氯甲烷(60ml)稀释并随后用饱和NaHCO3(2×50ml)和水(50ml)洗涤。随后将有机层进行干燥(MgSO4),过滤并减压下除去溶剂。粗产物通过快速色谱法(2%在二氯甲烷中的乙酸乙酯)进行纯化,这获得作为黄色粘性油的产物43(0.33g,0.12mmol,46%)。1H NMR(400MHz,CDCl3),δ=0.78(12H,t,J=7.1Hz,CH3),0.90(6H,t,J=6.9Hz,CH3),1.06-1.65(96H,m,CH2),1.72-1.89(16H,m,CH2),3.47-3.50(4H,m,CH2),4.00-4.05(12H,m,CH2),4.31(4H,t,J=6.7Hz,CH2),6.65(4H,s,=CH),6.99-7.01(8H,m,Ar-H),7.37(2H,s,Ar-H),7.57-7.59(8H,m,Ar-H),7.65-7.73(16H,m,Ar-H),7.78-7.84(8H,m,Ar-H),8.07(4H,br s,Ar-H);19F NMR(376MHz,CDCl3),δ=-108.93(CF2),-107.21(CF2);MS(MALDI+):m/z理论值[M+H]+=2799.5,实测值=2799.1。
方案8 具有其它交联剂结构的OLED材料的合成
上述化学由于合成的模块化性质而能够用于制备相关结构的替代OLED材料。例如,含有3型交联剂结构的化合物50能够由48制成。受保护的S3间隔单元48,以及其他类似的结构,都是用于合成适用于OLED中的材料的有用的中间体。示例性试剂提供如下。该化学能够同样适用于产生本文中公开的其它交联结构。
试剂和条件:i)Mg,THF,回流,1.5h;ii)Li2[CuCl4],THF,-78℃至室温,90h;iii)Br2,CH3CO2H,rt,1h;iv)5-己炔-1-醇,PdCl2(PPh3)2,NH(iPr)2,CuI,THF,rt,16h;v)TsCl,C2H5N,0℃至室温,16h;vi)Cs2CO3,DMF,90℃,18h;vii)Pd/C,H2,rt,20h;viii)4-溴-丁醇乙烯基醚,K2CO3,丁酮,回流,16h。
方案9 具有3型S3间隔单元的可交联氟烷基芴衍生物58的合成
4-(7-溴-2,1,3-苯并噻二唑-4-基)-苯酚51
将4-羟基苯基硼酸(3.50g,0.0254mol)、4,7-二溴苯并[c]-1,2,5-噻二唑(化合物7,10.0g,0.0340mol)、K2CO3(10.5g,0.0761mol)、二氧六环(150ml)和水(30ml)都加入到3-口圆底烧瓶中并实施三次冻融泵循环。随后,加入Pd(PPh3)4(1.47g,1.27mmol)并将反应混合物在90℃下搅拌2天。将反应混合物用10%v/v稀盐酸酸化并萃取至CH2Cl2(2×200ml)中。合并的有机层用水(2×200ml)洗涤,干燥(MgSO4),过滤并将粗产物通过重力柱层析(硅胶)使用梯度洗脱(10%在己烷中的乙酸乙酯/20%在己烷中的乙酸乙酯)进行纯化以获得作为黄色粉末的51(2.70g,35%)。1H NMR(400MHz,CDCl3):δ(ppm)5.19(br.s,1H),6.99(d,2H),7.52(d,1H),7.81(d,2H),7.90(d,1H)。
4-(7-溴-2,1,3-苯并噻二唑-4-基)-THP苯酚52
将化合物51、3,4-二氢-2H-吡喃、对甲苯磺酸和干CH2Cl2在室温下搅拌过夜(14h)。加入几滴三甲胺并使用旋转蒸发器除去CH2Cl2。粗产物通过快速柱层析(硅胶)使用梯度洗脱(30%在己烷中的CH2Cl2/50%在己烷中的CH2Cl2/100%CH2Cl2)进行纯化以获得黄色粉末52(1.50g,39%)。1H NMR(400MHz,CDCl3):δ(ppm)1.59-1.78(m,3H),1.88-1.93(m,2H),1.99-2.09(m,1H),3.61-3.66(m,1H),3.90-3.96(m,1H),5.52(t,1H),7.21(d,2H),7.52(d,1H),7.84(d,2H),7.89(d,1H)。
2,7-二(4,4,5,5-四甲基-1,3,2-二氧杂硼戊环-2-基)-9,9-二(1,1,2,2-四氟辛基)芴53
将2,7-二溴-9,9-二(1,1,2,2-四氟辛烷)芴(化合物4,1.50g,2.12mmol)、双戊酰二硼(1.24g,4.87mmol)、乙酸钾(1.25g,12.7mmol)、PdCl2(dppf)(0.17g,0.21mmol)和干二氧六环(50ml)加入到圆底烧瓶中。实施三次冻融泵循环并将反应混合物加热至90℃2天。将反应混合物进行过滤并使用旋转蒸发器除去二氧六环。粗产物通过快速柱层析(硅胶)使用10%在己烷中的乙酸乙酯作为洗脱剂进行纯化以获得作为白色粉末的53(0.73g,43%)。1H NMR(400MHz,CDCl3):δ(ppm)0.80(t,6H),1.02-1.21(m,20H),1.36(s,24H),7.76(d,2H),7.95(dd,2H),8.19(s,2H)。
化合物54
将化合物52(0.75g,1.92mmol)、化合物53(0.70g,0.87mmol)、K2CO3(0.60g,4.36mmol)、甲苯(20ml)和水(10ml)都加入到3-口圆底烧瓶中并实施三次冻融泵循环。随后,加入Pd(PPh3)4(0.10g,0.09mmol)并将反应混合物在90℃下搅拌24h。将反应混合物倾倒入添加有水(10ml)和甲苯(10ml)的分液漏斗中。有机层减压浓缩,随后使用甲苯共沸干燥。将粗产物通过重力柱层析(硅胶)使用梯度洗脱(50%在己烷中的CH2Cl2/100%CH2Cl2)进行纯化以获得黄色粉末(0.80g,91%)。在Suzuki交叉耦合之后,双THP保护的化合物通过在MeOH(50ml)和浓H2SO4(5ml)中回流2h立即保护,没有进行结构表征,以获得作为黄色粉末的54(0.75g,96%)。1H NMR(400MHz,CDCl3):δ(ppm)0.75(t,6H),1.04-1.27(m,20H),5.25(br.s,2H),7.03(d,4H),7.77(d,2H),7.87(d,2H),7.91(d,4H),7.98(d,2H),8.28(dd,2H),8.40(s,2H)。
化合物55
将碳酸钾(0.20g,1.46mmol)加入到化合物54(0.72g,0.73mmol)和1-溴辛烷(0.14g,0.73mmol)在丁酮(50ml)中的溶液中并在氮气氛下回流搅拌过夜(油浴温度=80℃,20h)。过滤掉盐,减压下除去丁酮并将粗产物通过重力柱层析(硅胶)使用20%在己烷中的乙酸乙酯进行纯化以获得作为黄色粉末的55(0.30g,38%)。1H NMR(400MHz,CDCl3):δ(ppm)0.75(t,6H),0.91(t,3H),1.05-1.69(m,30H),1.85(五重峰,2H),4.07(t,2H),5.01(br.s,1H),7.03(d,2H),7.09(d,2H),7.77(d,1H),7.79(d,1H),7.87(d,2H),7.90-7.99(m,6H),8.28(dd,2H),8.40(s,2H)。
化合物56
将碳酸铯(0.20g,0.62mmol)加入到化合物7(0.25g,0.23mmol)和2,5-二((6-溴己基)氧基)对苯二甲酸二乙酯(0.06g,0.10mmol)在干DMF(10ml)中的溶液中并在氮气氛下在100℃搅拌24h。过滤掉盐,高真空(油泵)下除去DMF并将粗产物通过重力柱层析(硅胶)使用30%在己烷中的乙酸乙酯进行纯化以获得作为黄色粉末的56(0.25g,93%)。1H NMR(400MHz,CDCl3):δ(ppm)0.75(t,12H),0.91(t,6H),1.06-1.89(m,86H),4.06(重叠三重峰,12H),4.39(五重峰,4H),7.09(d,8H),7.37(s,2H),7.78(d,4H),7.87(d,4H),7.93-7.99(m,12H),8.28(dd,4H),8.40(s,4H)。
化合物57
将氢氧化钠水溶液(1g在10ml H2O中)加入到化合物56(0.20g,0.08mmol)在THF(10ml)中的溶液中并在60℃下搅拌48h。将反应混合物使用10%v/v稀盐酸(10ml)酸化并萃取至CH2Cl2(2×50ml)中。将合并的有机层用水(2×50ml)洗涤,干燥(MgSO4),过滤并使用旋转蒸发器除去溶剂以获得作为黄色玻璃状固体的57(0.20g,100%)。1H NMR(400MHz,CDCl3):δ(ppm)0.75(t,12H),0.90(t,6H),1.06-2.03(m,80H),4.08(重叠三重峰,8H),4.34(t,4H),7.09(d,8H),7.77(d,2H),7.79(d,2H),7.87(dd,4H),7.90-7.98(m,14H),8.28(dd,4H),8.40(s,4H),11.1(br.s,2H)。
化合物58
在氮气氛下一次性将DCC(0.08g,1.16mmol)室温下加入到化合物57(0.20g,0.08mmol),1,4-丁二醇乙烯基醚(0.09g,0.80mmol)和DMAP(4mg,0.03mmol)在干CH2Cl2(10ml)中的溶液中。将反应混合物室温搅拌20h并减压除去CH2Cl2。获得的残余物通过快速柱层析(硅胶)以梯度洗脱(10%在己烷中的乙酸乙酯/20%在己烷中的乙酸乙酯)进行纯化以获得作为玻璃状绿色固体的58(0.10g,46%)。1H NMR(400MHz,CDCl3):δ(ppm)0.75(t,12H),0.91(t,6H),1.06-1.89(m,88H),3.75(t,4H),3.99(dd,2H),4.06(重叠三重峰,12H),4.18(dd,2H),4.37(t,4H),6.47(dd,2H),7.09(d,8H),7.38(s,2H),7.78(d,4H),7.86(d,4H),7.93-7.99(m,12H),8.28(dd,4H),8.40(s,4H)。质量(MALDI)=2755.6(M+)。
- 还没有人留言评论。精彩留言会获得点赞!