一种合成氨基甲酸肟酯衍生物的新方法与流程
本发明属于农药化工合成技术领域,特别涉及一种由芳基酮肟、异腈、水合成氨基甲酸肟酯衍生物的新方法。
背景技术:
氨基甲酸肟酯类的化合物具有十分广泛的生物农药活性,是很多生物活性分子及农药分子的重要结构单元。为此,科研工作者对这类化合物进行了大量的研究,发现氨基甲酸肟酯类化合物具有抑制细菌,杀虫以及作为酶的抑制剂等重要作用。虽然此类化合物具有如此重要的应用,但是遗憾的是氨基甲酸肟酯类化合物的合成方法并不多。
构建氨基甲酸肟酯类衍生物的传统合成方法一般要用到肟和异氰酸酯(S.Y.Sita,C.M.Conway,K.Xie,R.Bertekap,C.Bourin,K.D.Burris,Bioorg.Med.Chem.Lett.20(2010)1272–1277.V.S.Georgiev,G.A.Bennett,L.A.Radov,D.K.Kamp,Arch.Pharm.(Weinheim)320.465-470(1987).C.Saturninoa,S.Petrosino,A.Ligresti,C.Palladino,G.D.Martino,T Bisogno,V.D.Marzo,and L.A.Trusso,Bioorganic&Medicinal Chemistry Letters 20(2010)1210–1213)。此类方法最大的问题在于采用较难合成的异氰酸酯为反应原料,步骤经济型差;除此之外,对于若干官能团兼容性差也是实际存在的难题。因此,发展环境友好的构建多种氨基甲酸肟酯类衍生物的合成方法一直受到科学界及工业界的广泛关注。
在有机合成化学中,异腈是一种非常有研究意义、实用价值的有机合成砌块。近年来,基于异腈参与的有机反应受到广泛关注(T.Hirai,L.-B.Han,J.Am.Chem.Soc.2006,128,7422;H.Jiang,B.Liu,Y.Li,A.Wang,H.Huang,Org.Lett.2011,13,1028-1031;F.Zhou,K.Ding,Q.Cai,Chem.Eur.J.2011,17,12268;H.Jiang,B.Liu,Y.Li,A.Wang,H.Huang,Org.Lett.2011,13,1028;T.Nanjo,C.Tsukano,Y.Takemoto,Org.Lett.2012,14,4270;T.Vlaar,R.C.Cioc,P.Mampuys,B.U.W.Maes,R.V.A.Orru,E.Ruijter,Angew.Chem.2012,124,13235;Angew.Chem.Int.Ed.2012,51,13058;L.D.Miranda,E.H.-Va′zquez′J.Org.Chem.2015,80,10611-10623;J.Liu,Z.Liu,P.Liao,L.Zhang,T.Tu,and X.Bi,Angew.Chem.Int.Ed.2015,54,1–6)。但是目前还没有利用异腈为原料直接合成氨基甲酸肟酯类衍生物的报道。
技术实现要素:
为了克服上述现有技术的缺点与不足,本发明的首要目的在于提供一种合成氨基甲酸肟酯衍生物的新方法。该方法以芳基酮肟、异腈和水为原料,在钯盐的催化下,发生串联反应,一步合成氨基甲酸肟酯衍生物。该方法的反应原料简单易得,反应操作安全简便,反应过程环境友好,底物适用性广,官能团兼容性强,具有潜在的实用价值。
本发明的目的通过下述方案实现:
一种合成氨基甲酸肟酯衍生物的新方法,具体包括以下步骤:
在反应瓶中,加入芳基酮肟、异腈和水为原料,在钯盐为催化剂、碱作为添加剂,有机溶剂为溶剂的条件下加热反应,反应结束后将所得反应液冷却至室温然后纯化,即得所述的氨基甲酸肟酯衍生物。
其反应如下式所示:
所述的加热反应是指在80~100℃下搅拌反应3~12h。
所述的芳基酮肟可为苯乙酮肟、4-氟苯乙酮肟、4-氯苯乙酮肟、4-腈基苯乙酮肟、3-氯苯乙酮肟、2-氯苯乙酮肟、2-萘乙酮肟、3,4-二甲氧基苯乙酮肟、苯丙酮肟、苯并环己酮肟、6-甲氧基苯并环己酮肟、苯并环庚酮肟中的至少一种。
所述的异氰可为叔丁基异腈、正丁基异腈、1,1,3,3-四甲基丁基异腈、环戊基异腈、环己基异腈、1-异腈基金刚烷、对甲氧基苯基异腈中的至少一种。
所用的芳基酮肟、异腈和水的摩尔比为1:1:(5~10)。
优选的,所用的芳基酮肟、异腈和水的摩尔比为1:1:5。
所述的钯盐可为四三苯基膦钯,三(二亚苄基丙酮)二钯、双三苯基磷二氯化钯中的至少一种。
所用的钯盐与芳基酮肟的摩尔比为(0.01-0.05):1。
优选的,所用的钯盐与芳基酮肟的摩尔比为0.03:1。
所述的添加剂为乙酸钠或碳酸铯。
所述的有机溶剂可为甲苯、N,N-二甲基甲酰胺、乙腈和1,2-二氯乙烷中的至少一种。
所述的纯化是指将反应液过滤,然后减压蒸馏除去溶剂以得到粗产物,再将粗产物经柱层析分离纯化。
所述的柱层析中,所用的洗脱液为石油醚和乙酸乙酯的混合溶剂;石油醚和乙酸乙酯之间的比例范围值为(10~200):1。
所得的氨基甲酸肟酯衍生物为顺式构型(Z)。
本发明的机理为:以芳基肟、异腈、水为反应原料,以钯盐作为催化剂,以碱作为添加剂的作用下一步合成取代的氨基甲酸肟酯。现有的直接合成氨基甲酸肟脂主要采用肟和异氰酸酯的偶联反应在无金属条件下来实现,其他类型的合成方法并不常见。而利用异腈和肟为原料直接合成氨基甲酸肟脂类衍生物是采用了钯催化的有机合成的方法,在钯催化剂的作用下活化肟的氧氢键,然后插入异腈,在水作为亲核试剂的条件下得到氨基甲酸肟脂,异腈可以作为酰胺基团的来源。这样的方法的得益于过渡金属催化有机合成领域的发展,使我们可以从中汲取新的思想发展新的合成方法。
本发明相对于现有技术,具有如下的优点及有益效果:
本发明的合成氨基甲酸肟酯的新方法。反应原料简单易得,反应操作安全简便,反应过程环境友好,底物适用性广,官能团兼容性强,原子经济性好。该方法在农药、医药中具有广泛用途。
附图说明
图1为实施例1-9所得产物的核磁共振氢谱图。
图2为实施例1-9所得产物的核磁共振碳谱图。
图3为实施例10所得产物的核磁共振氢谱图。
图4为实施例10所得产物的核磁共振碳谱图。
图5为实施例11所得产物的核磁共振氢谱图。
图6为实施例11所得产物的核磁共振碳谱图。
图7为实施例12所得产物的核磁共振氢谱图。
图8为实施例12所得产物的核磁共振碳谱图。
图9为实施例13所得产物的核磁共振氢谱图。
图10为实施例13所得产物的核磁共振碳谱图。
图11为实施例14所得产物的核磁共振氢谱图。
图12为实施例14所得产物的核磁共振碳谱图。
图13为实施例15所得产物的核磁共振氢谱图。
图14为实施例15所得产物的核磁共振碳谱图。
图15为实施例16所得产物的核磁共振氢谱图。
图16为实施例16所得产物的核磁共振碳谱图。
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例中所用试剂均可从市场常规购得。
实施例1
在25毫升试管中加入0.2毫摩尔苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的三(二亚苄基丙酮)二钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应12小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为21%。所得产物的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示,说明成功合成了产物。
实施例2
在25毫升试管中加入0.2毫摩尔l苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的双三苯基磷二氯化钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应12小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为33%。所得产物的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示,说明成功合成了产物。
实施例3
在25毫升试管中加入0.2毫摩尔苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2mL甲苯作为溶剂,在80℃条件下搅拌反应12小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为68%。所得产物的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示,说明成功合成了产物。
实施例4
在25毫升试管中加入0.2毫摩尔苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在90℃条件下搅拌反应12小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为54%。所得产物的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示,说明成功合成了产物。
实施例5
在25毫升试管中加入0.2毫摩尔苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在100℃条件下搅拌反应12小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为40%。所得产物的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示,说明成功合成了产物。
实施例6
在25毫升试管中加入0.2毫摩尔苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应6小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为67%。所得产物的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示,说明成功合成了产物。
实施例7
在25毫升试管中加入0.2毫摩尔苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的碳酸铯,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为18%。所得产物的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示,说明成功合成了产物。
实施例8
在25毫升试管中加入0.2毫摩尔苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升N,N-二甲基甲酰胺(DMF)作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为20%。所得产物的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示,说明成功合成了产物。
实施例9
在25毫升试管中加入0.2毫摩尔苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤,然后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,产率为66%。其中所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂。
所得产物的结构表征数据如下所示:
IR(KBr):2969,2364,1750,1652,1457,1317,1257,1051,938cm-1
1H NMR(400MHz,CDCl3)δ7.65(dd,J=7.8,1.5Hz,2H),7.43(t,J=7.3Hz,3H),6.35(s,1H),2.40(s,3H),1.41(s,9H),如图1所示。
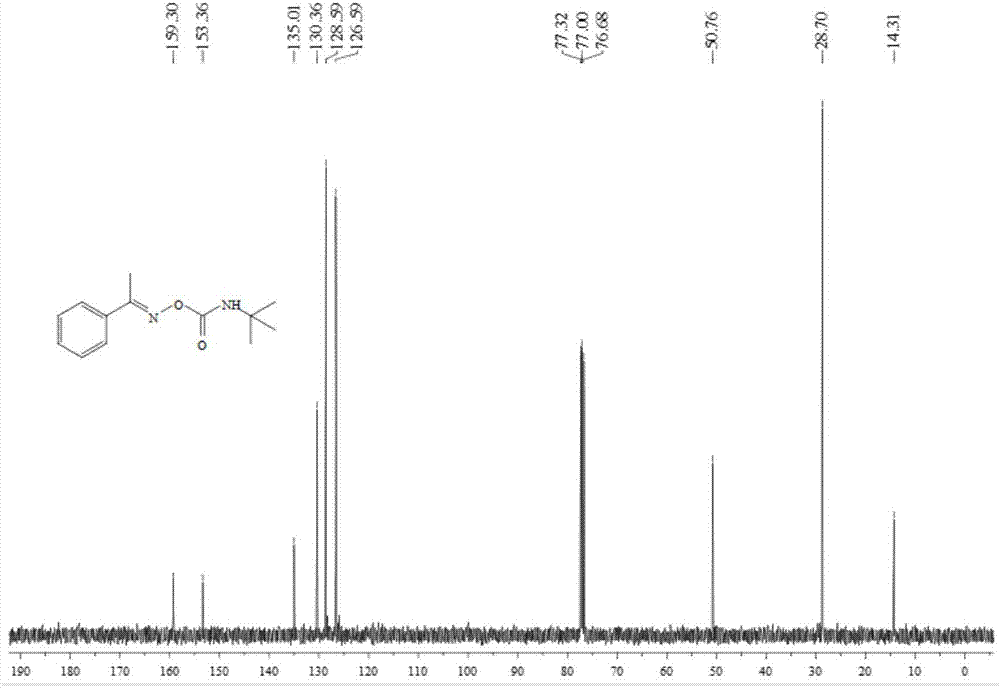
13C NMR(100MHz,CDCl3)δ159.3,153.4,135.0,130.4,128.6,126.6,50.8,28.7,14.3ppm,如图2所示。
ESI-HRMS:C13H18N2NaO2[M+Na]+计算值:257.1260,实验值:257.1262.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例10
在25毫升试管中加入0.2毫摩尔4-氟苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为54%。
所得产物的结构表征数据如下所示:
IR(KBr):2970,1749,1676,1457,1318,1262,1051,940cm-1
1H NMR(400MHz,CDCl3)δ7.66(dd,J=7.8,5.6Hz,2H),7.10(t,J=8.3Hz,2H),6.25(s,1H),2.39(s,3H),1.41(s,9H),如图3所示。
13C NMR(100MHz,CDCl3)δ165.3,162.8,158.3,153.2,131.1,128.7,128.6,115.8,115.6,50.9,28.7,14.4ppm,如图4所示。
ESI-HRMS:C13H17FN2NaO2[M+Na]+计算值:275.1166,实验值:275.1169.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例11
在25毫升试管中加入0.2毫摩尔4-氯苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为52%。
所得产物的结构表征数据如下所示:
IR(KBr):2981,1748,1642,1452,1370,1183,1048,937cm-1
1H NMR(400MHz,CDCl3)δ7.68(d,J=8.2Hz,2H),7.26(d,J=8.3Hz,2H),5.20(s,1H),2.26(s,3H),1.33(s,9H),如图5所示。
13C NMR(100MHz,CDCl3)δ167.1,163.0,137.4,136.1,128.6,128.4,51.0,28.8,18.0ppm,如图6所示。
ESI-HRMS:C13H17ClN2NaO2[M+Na]+计算值:291.0871,实验值:291.0869.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例12
在25毫升试管中加入0.2毫摩尔4-腈基苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为63%。
所得产物的结构表征数据如下所示:
IR(KBr):2971,1742,1499,1368,1261,1050,940cm-1
1H NMR(400MHz,CDCl3)δ7.78(d,J=8.3Hz,2H),7.71(d,J=8.4Hz,2H),6.11(s,1H),2.41(s,3H),1.41(s,9H),如图7所示。
13C NMR(100MHz,CDCl3)δ157.8,152.7,139.3,132.4,127.3,118.1,113.9,51.1,28.7,14.2ppm,如图8所示。
ESI-HRMS:C14H17N3NaO2[M+Na]+计算值:282.1213,实验值:282.1211.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例13
在25毫升试管中加入0.2毫摩尔的3-氯苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为65%。
所得产物的结构表征数据如下所示:
IR(KBr):2970,1749,1620,1457,1367,1261,1051,943cm-1
1H NMR(400MHz,CDCl3)δ7.64(s,1H),7.54(d,J=7.7Hz,1H),7.43(d,J=8.1Hz,1H),7.36(t,J=7.8Hz,1H),6.22(s,1H),2.39(s,3H),1.42(s,9H),如图9所示。
13C NMR(100MHz,CDCl3)δ158.2,153.0,136.9,134.7,130.4,129.9,126.8,124.8,51.0,28.7,14.4ppm,如图10所示。
ESI-HRMS:C13H17ClN2NaO2[M+Na]+计算值:291.0871,实验值:291.0868.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例14
在25毫升试管中加入0.2毫摩尔的2-氯苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为69%。
所得产物的结构表征数据如下所示:
IR(KBr):2969,1751,1627,1457,1366,1261,1049,936cm-1
1H NMR(400MHz,CDCl3)δ7.44(d,J=7.6Hz,1H),7.35(dt,J=14.0,6.8Hz,3H),6.23(s,1H),2.38(s,3H),1.38(s,9H),如图11所示。
13C NMR(100MHz,CDCl3)δ160.3,153.1,135.4,132.5,130.6,130.2,130.0,126.9,50.9,28.7,17.7ppm,如图12所示。
ESI-HRMS:C13H17ClN2NaO2[M+Na]+计算值:291.0871,实验值:291.0868.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例15
在25毫升试管中加入0.2毫摩尔的β-萘乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为57%。
所得产物的结构表征数据如下所示:
IR(KBr):2959,2360,2338,1740,1646,1469,1364,1260,1052,954cm-1
1H NMR(400MHz,CDCl3)δ8.09(s,1H),7.86(s,4H),7.57–7.52(m,2H),6.38(s,1H),2.53(s,3H),1.44(s,9H),如图13所示。
13C NMR(100MHz,CDCl3)δ159.2,153.4,134.2,132.9,132.3,128.6,128.4,127.7,127.3,127.3,126.7,123.2,50.9,28.8,14.2ppm,如图14所示。
ESI-HRMS:C17H21N2O2[M+H]+计算值:285.1598,实验值:285.1598.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例16
在25毫升试管中加入0.2毫摩尔的3,4-二甲氧基苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为60%。
所得产物的结构表征数据如下所示:
IR(KBr):2968,1745,1498,1457,1368,1148,943cm-1
1H NMR(400MHz,CDCl3)δ7.16(d,J=8.8Hz,2H),6.82(d,J=8.2Hz,1H),6.23(s,1H),3.85(s,6H),2.31(s,3H),1.34(s,9H),如图15所示。
13C NMR(100MHz,CDCl3)δ158.9,153.5,151.1,148.9,127.5,120.2,110.7,109.1,55.9,55.9,50.7,28.7,14.1,如图16所示。
ESI-HRMS:C15H22N2O4[M+Na]+计算值:317.1472,实验值:317.1474。
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例17
在25毫升试管中加入0.2毫摩尔的苯丙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4mmol的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为75%。
所得产物的结构表征数据如下所示:
IR(KBr):2972,1748,1638,1458,1366,1261,1053,947cm-1
1H NMR(400MHz,CDCl3)δ7.64(dd,J=7.8,1.7Hz,2H),7.44(ddd,J=7.7,5.4,2.2Hz,3H),6.37(s,1H),2.90(q,J=7.6Hz,2H),1.41(s,9H),1.18(t,J=7.6Hz,3H)。
13C NMR(100MHz,CDCl3)δ164.2,153.5,134.0,130.3,128.7,126.9,50.8,28.8,21.5,11.2ppm。
ESI-HRMS:C14H20N2NaO2[M+Na]+计算值:271.1417,实验值:271.1422.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例18
在25毫升试管中加入0.2毫摩尔的苯并环己酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为70%。
所得产物的结构表征数据如下所示:
IR(KBr):2968,1749,1620,1456,1366,1261,1053,944cm-1.
1H NMR(400MHz,CDCl3)δ7.93(d,J=7.8Hz,1H),7.34(t,J=7.2Hz,1H),7.24(t,J=7.5Hz,1H),7.18(d,J=7.6Hz,1H),6.41(s,1H),2.91(t,J=6.6Hz,2H),2.80–2.73(m,2H),1.91–1.83(m,2H),1.43(s,9H)。
13C NMR(100MHz,CDCl3)δ158.2,153.4,141.0,130.4,128.9,128.8,126.4,124.5,50.7,29.4,28.7,25.4,21.1ppm。
ESI-HRMS:C15H20N2NaO2[M+Na]+计算值:283.1417,实验值:283.1422.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例19
在25毫升试管中加入0.2毫摩尔的6-甲氧基苯并环己酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为73%。
所得产物的结构表征数据如下所示:
IR(KBr):2966,1741,1619,1457,1355,1258,1053,946cm-1
1H NMR(400MHz,CDCl3)δ7.87(d,J=8.8Hz,1H),6.78(d,J=8.7Hz,1H),6.67(s,1H),6.40(s,1H),3.82(s,3H),2.87(t,J=6.2Hz,2H),2.76–2.70(m,2H),1.89–1.81(m,2H),1.41(s,9H)。
13C NMR(100MHz,CDCl3)δ161.3,158.0,153.7,143.0,126.3,121.6,113.3,112.9,55.3,50.7,29.9,28.8,25.4,21.2ppm。
ESI-HRMS:C16H22N2NaO3[M+Na]+计算值:313.1523,实验值:313.1527.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例20
在25毫升试管中加入0.2毫摩尔的苯并环庚酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为73%。
所得产物的结构表征数据如下所示:
IR(KBr):2966,1743,1646,1456,1365,1259,1052,937cm-1
1H NMR(400MHz,CDCl3)δ7.38(dd,J=20.3,7.3Hz,2H),7.28(d,J=8.0Hz,1H),7.18(d,J=7.1Hz,1H),6.39(s,1H),2.88–2.80(m,2H),2.77(d,J=5.3Hz,2H),1.83–1.75(m,2H),1.66(d,J=4.8Hz,2H),1.39(s,9H)。
13C NMR(100MHz,CDCl3)δ166.5,153.5,139.8,134.7,130.2,129.0,127.7,126.5,50.7,31.8,28.7,27.7,25.7,21.6ppm。
ESI-HRMS:C16H22N2NaO2[M+Na]+计算值:297.1573,实验值:297.1571.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例21
在25毫升试管中加入0.2毫摩尔的苯乙酮肟、0.2毫摩尔的叔丁烷异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为77%。
所得产物的结构表征数据如下所示:
IR(KBr):2958,1741,1624,1445,1373,1239,1158,1042,991,929,852,762,693cm-1
1H NMR(400MHz,CDCl3)δ7.66(dd,J=8.0,1.5Hz,2H),7.47–7.40(m,3H),6.45(s,1H),3.33(dd,J=13.2,7.1Hz,2H),2.42(s,3H),1.57(dt,J=14.9,7.5Hz,2H),1.38(dd,J=15.1,7.4Hz,2H),0.94(t,J=7.3Hz,3H)
13C NMR(100MHz,CDCl3)δ159.9,155.5,134.9,130.5,128.6,126.7,40.9,31.8,19.9,14.4,13.7ppm
ESI-HRMS:C13H18N2NaO2[M+Na]+计算值:257.1260,实验值:257.1264.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例22
在25毫升试管中加入0.2毫摩尔的苯乙酮肟、0.2毫摩尔的1,1,3,3-四甲基丁基异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为73%。
所得产物的结构表征数据如下所示:
IR(KBr):2954,1743,1680,1573,1447,1367,1252,1157,1081,991,937,783cm-1
1H NMR(400MHz,CDCl3)δ7.67–7.63(m,2H),7.44(t,J=7.7Hz,3H),6.52(s,1H),2.41(s,3H),1.75(s,2H),1.47(s,6H),1.04(s,9H)
13C NMR(100MHz,CDCl3)δ159.2,153.1,135.1,130.4,128.6,126.6,54.6,52.3,31.6,31.5,29.0,14.4ppm
ESI-HRMS:C17H26N2NaO2[M+Na]+计算值:313.1886,实验值:313.1893.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例23
在25毫升试管中加入0.2毫摩尔的苯乙酮肟、0.2毫摩尔的环戊异腈、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为60%。
所得产物的结构表征数据如下所示:
IR(KBr):2958,1741,1621,1573,1447,1370,1229,1059,992,911,784cm-1
1H NMR(400MHz,CDCl3)δ7.65(dd,J=7.9,1.5Hz,2H),7.49–7.41(m,3H),6.34(d,J=5.7Hz,1H),4.18–4.09(m,1H),2.42(s,3H),2.04(td,J=11.8,6.3Hz,2H),1.75–1.68(m,2H),1.66–1.58(m,2H),1.54–1.46(m,2H)
13C NMR(100MHz,CDCl3)δ159.9,154.9,135.0,130.5,128.6,126.7,52.9,33.1,23.5,14.5ppm
ESI-HRMS:C14H18N2NaO2[M+Na]+计算值:269.1260,实验值:269.1264.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例24
在25毫升试管中加入0.2毫摩尔的苯乙酮肟、0.2毫摩尔的环己异氰、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为67%。
所得产物的结构表征数据如下所示:
IR(KBr):2925,2853,1755,1496,1373,1318,1055,965cm-1
1H NMR(400MHz,CDCl3)δ7.69–7.63(m,2H),7.48–7.40(m,3H),6.33(d,J=6.8Hz,1H),3.72–3.63(m,1H),2.42(s,3H),2.02(dd,J=12.1,3.0Hz,2H),1.76–1.59(m,4H),1.42–1.34(m,2H),1.25(s,2H)
13C NMR(100MHz,CDCl3)δ159.8,154.6,135.0,130.5,128.7,126.7,50.0,33.2,25.5,24.8,14.5ppm
ESI-HRMS:C15H20N2NaO2[M+Na]+计算值:283.1417,实验值:218.1418.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例25
在25毫升试管中加入0.2毫摩尔的苯乙酮肟、0.2毫摩尔的1-异氰基金刚烷、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为75%。
所得产物的结构表征数据如下所示:
IR(KBr):2909,2852,1748,1452,1365,1275,1138,1048,974,928,761cm-1.
1H NMR(400MHz,CDCl3)δ7.66(dd,J=8.0,1.6Hz,2H),7.46–7.39(m,3H),6.29(s,1H),2.41(s,3H),2.11(s,3H),2.04(d,J=2.7Hz,6H),1.70(s,6H)
13C NMR(100MHz,CDCl3)δ159.1,152.9,135.1,130.4,128.6,126.6,51.2,41.6,36.3,29.4,14.4
ESI-HRMS:C19H24N2NaO2[M+Na]+:计算值:335.1730,实验值:335.1730。
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
实施例26
在25毫升试管中加入0.2毫摩尔的苯乙酮肟、0.2毫摩尔的对甲氧基苯基异氰、1毫摩尔的水、0.006毫摩尔的四三苯基磷钯和0.4毫摩尔的乙酸钠,加入2毫升甲苯作为溶剂,在80℃条件下搅拌反应3小时后,停止加热及搅拌,冷却至室温,过滤后加入乙酸乙酯萃取三遍,硫酸镁干燥,减压旋蒸除去溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为20:1的石油醚:乙酸乙酯混合溶剂,产率为58%。
所得产物的结构表征数据如下所示:
IR(KBr):2929,2836,1735,1599,1413,1372,1245,1177,1007,913cm-1
1H NMR(400MHz,CDCl3)δ8.27(s,1H),7.71(d,J=6.6Hz,2H),7.51–7.40(m,5H),6.89(d,J=8.9Hz,2H),3.80(s,3H),2.49(s,3H)
13C NMR(100MHz,CDCl3)δ160.6,156.6,152.7,134.7,130.7,130.0,128.7,126.8,121.8,114.3,55.5,14.7ppm
ESI-HRMS:C16H16N2NaO3[M+Na]+计算值:307.1053,实验值:307.1057.
根据以上数据推断所得产物得结构如下所示:说明成功合成了目标产物。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!