高纯度12‑甲基十三醛的制备方法与流程
本发明涉及精细化工领域,具体涉及一种高纯度12-甲基十三醛的制备方法。
背景技术:
12-甲基十三醛是近年来新兴的一种咸味香精的单体原料,该化合物天然存在于炖牛肉汁、羔羊、马鹿、鸡和火鸡中,可应用于食品加工。其具有牛脂香气、油炸食品的头香、鸡肉油脂香,在高度稀释的情况下,依然香气馥郁;同时,因其分子量较大,沸点较高等特点,该化合物可广泛应用于耐高温及留香时间长的咸味香精中,也可以应用于高档水果香精中。
由于其巨大的经济价值和应用价值,其人工合成工艺也在被化学家不断地探索之中,国内文献,对于该化合物合成方法的记载较少。目前,国内外总结起来,大概有以下几种方法:
方法一:
以癸二酸为原料,制得癸二酸单酰氯乙酯,无需进行产物分离,继续与溴代异丁基镁反应,然后经锌汞齐还原得到12-甲基十三酸(60~65%),再酯化、还原,得到目标产物12-甲基十三醛;
此方法原料易获取,但工艺路线较长,且使用锌汞齐等原料,操作复杂且锌汞齐为剧毒,对实验人员危害较大,并不适用于工业化生产;
方法二:
以4-甲基戊醛和8-溴代辛酸为原料,经鏻盐的witting反应和加氢后,制得12-甲基十三酸,再经酯化还原制得最终产品;
此方法原料难获取,工艺复杂,工艺中使用正丁基锂,正丁基锂属于易燃危险试剂,增加实验操作的危险性,工业化难度大;
方法三:11-溴代十一醇和丙酮为原料,经鏻盐的witting反应和加氢后,氧化制得12-甲基十三醛;
方法四:
以11-溴代-十一-烯-1为原料,经与溴代异丙基镁反应,再经有机硼烷的双氧水氧化制得12-甲基十三醇(85%),继而经吡啶铬酸鎓盐氧化制得产品(84%);
方法三和方法四原料昂贵,反应步骤长,方法三中使用正丁基锂,正丁基锂属于易燃危险试剂,危险性高,方法四中利用醋酸汞为反应试剂,容易造成重金属污染,三废较大。
技术实现要素:
本发明要解决的技术问题是,提供一种高纯度12-甲基十三醛的制备方法,其实现了12-甲基十三醛的人工合成,该方法原料成本低且易获取,步骤少,反应条件温和,杂质含量低且易纯化,反应重复性好且稳定性高,适合工业化大规模生产。
一种高纯度12-甲基十三醛的制备方法,其以10—十一烯酸为原料,经过酰胺化反应、格氏反应、氢化还原和烯基臭氧化四步反应,并得到最终产品,具体操作如下:
步骤一酰胺化反应
室温下,向反应釜中加入二氯甲烷、10-十一烯酸,搅拌下滴加氯化亚砜,滴毕升温至T1反应,检测反应完成,浓缩,得到A;
室温下,向反应釜中一次加入二氯甲烷、N,O-二甲基羟胺盐酸盐,降温至T2,滴加A的二氯甲烷溶液,滴毕,10~40℃反应;
GC检测反应,反应完成向反应釜中滴加质量分数W1的稀盐酸,充分搅拌20~60min,分液,水相用二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得B;
步骤二格氏反应
向反应釜中依次加入四氢呋喃和B,体系降温至T3,滴加摩尔浓度为M1的异丁基溴化镁,滴加完毕,升至10~50℃反应;
GC检测反应,反应完成,体系降温至T4,向其中滴加饱和氯化铵水溶液,用二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得到C;
步骤三氢化还原
向四口瓶中依次加入二甘醇、氢氧化钾,T5温度下,待氢氧化钾溶解后,加入C,剧烈搅拌下滴加水合肼,滴毕,T6温度下反应2~3h后,T7温度下继续反应;
GC检测反应完成,体系降至室温,向其中加入水,用正己烷萃取,无水硫酸钠干燥,浓缩,得粗品,减压蒸馏,得到D;
步骤四烯基臭氧化
向四口瓶中依次加入二氯甲烷和D,降温至T8,剧烈搅拌下,向反应釜中通入臭氧,GC检测反应完成,停止通入臭氧,缓慢滴加二甲基硫醚,无剧烈升温后停止滴入,体系降至室温,加入水,分液,二氯甲烷相用无水硫酸钠干燥,浓缩,得液体产品TM。
作为本技术方案的进一步改进,步骤一中:
10-十一烯酸:二氯甲烷(质量体积比)为1g:16~25ml;
10-十一烯酸:氯化亚砜(摩尔比)为1:2~4;
所述T1为20~40℃;
所述T2为0~5℃;
所述W1为5~20wt%;
N,O-二甲基羟胺盐酸盐:二氯甲烷(质量体积比)为1g:10~20ml;
10-十一烯酸:N,O-二甲基羟胺盐酸盐(摩尔比)为1:1.1~1.3;
10-十一烯酸:稀盐酸溶液(质量体积比)为1g:3~8ml。
作为本技术方案的进一步改进,步骤二中:
所述T3为0~25℃;
所述M1为1mol/L或2mol/L;
所述T4为-10~20℃;
B:四氢呋喃(质量体积比)为1g:3~5ml;
B:异丁基溴化镁(摩尔比)为1:1.1~1.3;
B:饱和氯化铵水溶液(质量体积比)为1g:2~5ml。
作为本技术方案的进一步改进,步骤三中:
所述T5为70~90℃;
所述T6为110~130℃;
所述T7为190~210℃;
C:二甘醇(质量体积比)为1g:4~6ml;
C:氢氧化钾(摩尔比)为1:1.3~1.5;
C:水合肼(摩尔比)1:2.1~2.5。
作为本技术方案的进一步改进,步骤三中:
减压蒸馏收集的馏分为118℃/20Pa的馏分。
作为本技术方案的进一步改进,步骤四中:
分子D:二氯甲烷(质量体积比)为1g:5~8ml。
本发明采用上述技术方案,带来的有益效果是:
本发明采用10-十一酸为起始原料,成本低且以获取,降低生产成本;所述高纯度12-甲基十三醛的制备方法,各反应步骤后处理简单,收率和纯度均较高;所述高纯度12-甲基十三醛的制备方法,各反应步骤反应条件温和,后处理简单,适合工业化大规模生产;本发明解决了长期以来困扰12-甲基十三醛合成中,原材料昂贵、纯化难、工艺复杂、生产危险性高等问题,很好的实现了12-甲基十三醛的人工合成,为国内合成和使用提供了便利,带动更大的经济效益产生。
附图说明
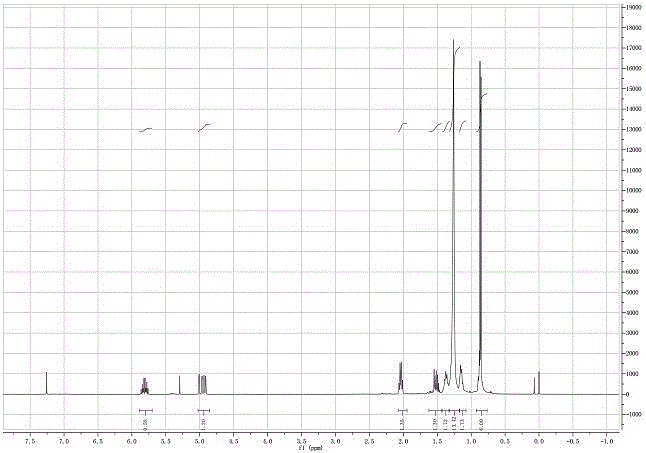
图1 本发明12-甲基十三醛气相质谱图;
图2 本发明12-甲基十三醛核磁图。
具体实施例
为了更好的理解本发明技术方案的内容,以下为本发明的具体实施例,从详细的投料和反应操作角度对本发明技术方案做进一步说明,但是并不作为本发明技术方案的保护范围的限定,需要指出的是,一切在不脱离本发明的发明精神的前提下,对于本发明技术方案中描述内容的替换或者改进,例如,对于反应物投料量的变换、反应条件的改良等,均应落于本发明技术方案的保护范围之内;
所述高纯度12-甲基十三醛的制备方法的合成路线如下:
所述高纯度12-甲基十三醛的制备方法包括以下步骤:
一种高纯度12-甲基十三醛的制备方法,其以10—十一烯酸为原料,经过酰胺化反应、格氏反应、氢化还原和烯基臭氧化四步反应,并得到最终产品,具体操作如下:
步骤一酰胺化反应
室温下,向反应釜中加入二氯甲烷、10-十一烯酸,搅拌下滴加氯化亚砜,滴毕,10~40℃反应,检测反应完成,浓缩,得到A;10-十一烯酸与二氯甲烷的质量体积比为1g:16~25ml;10-十一烯酸与氯化亚砜的摩尔比为1:2~4;
室温下,向反应釜中加入二氯甲烷和N,O-二甲基羟胺盐酸盐,降温至0~5℃,滴加A的二氯甲烷溶液,滴毕,10~40℃反应;N,O-二甲基羟胺盐酸盐与二氯甲烷的质量体积比为1g:10~20ml;10-十一烯酸与N,O-二甲基羟胺盐酸盐的摩尔比为1:1.1~1.3;
GC检测反应,反应完成向反应釜中滴加5~20wt%的稀盐酸,10-十一烯酸与稀盐酸溶液的质量体积比为1g:3~8ml,充分搅拌20~60min,分液,水相用二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得到B;
步骤二格氏反应
向反应釜中依次加入四氢呋喃、分子B,体系降温至0~25℃,滴加摩尔浓度为1mol/L或2mol/L 的异丁基溴化镁,滴加完毕,10~50℃反应;B与四氢呋喃的质量体积比为1g:3~5ml;B与异丁基溴化镁的摩尔比为1:1.1~1.3;
GC检测反应,反应完成,体系降温至-10~20℃,向其中滴加饱和氯化铵水溶液,B与饱和氯化铵水溶液的质量体积比为1g:2~5ml,用二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得到C;
步骤三氢化还原
向反应釜中依次加入二甘醇和氢氧化钾,70~90℃温度下,待氢氧化钾溶解后,加入C,剧烈搅拌下滴加水合肼,滴毕,110~130℃温度下反应2~3h后,升温至190~210℃继续反应;C与二甘醇的质量体积比为1g:4~6ml;C与氢氧化钾的摩尔比为1:1.3~1.5;C与水合肼的摩尔比1:2.1~2.5;
GC检测反应完成,体系降至室温,向其中加入水,用正己烷萃取,无水硫酸钠干燥,浓缩,得粗品,减压蒸馏收集118℃/20Pa的馏分,得到D;
步骤四烯基臭氧化
向四口瓶中依次加入二氯甲烷和D,D与二氯甲烷的质量体积比为1g:5~8ml,
降温至-50~-70℃,剧烈搅拌下,向反应釜中通入臭氧,GC检测反应完成,停止通入臭氧,缓慢滴加二甲基硫醚,无剧烈升温后停止滴入,体系温度10~30℃时,加入水,分液,二氯甲烷相用无水硫酸钠干燥,浓缩,得液体产品TM。
实施例一
步骤一酰胺化反应
室温下,向反应釜中加入800ml二氯甲烷和50g 10-十一烯酸,搅拌下滴加64.6g氯化亚砜,滴毕升温至40℃反应,检测反应完成,浓缩,得到A;
室温下,向反应釜中加入291ml二氯甲烷和29.1g N,O-二甲基羟胺盐酸盐,降温至0℃,滴加分子A的二氯甲烷溶液,滴完升至40℃反应;
GC检测反应,反应完成向反应釜中滴加质量分数20%的稀盐酸150ml,充分搅拌20~60min,分液,水相分批用270ml二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得63.3g B,气相质谱纯度95.0%,收率97.5%;
步骤二格氏反应
向反应釜中依次加入四氢呋喃190ml和B 63.3g,体系降温至0℃,滴加摩尔浓度为2mol/L的异丁基溴化镁146ml,滴加完毕,升至50℃反应;
GC检测反应,反应完成,体系降温至0℃,向其中滴加饱和氯化铵水溶液120ml,分批用260ml二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得59.0 g C,气相质谱纯度90.5%,收率90.0%;
步骤三氢化还原
向反应釜中依次加入二甘醇214ml和氢氧化钾17.4 g,升温至90℃,待氢氧化钾溶解后,加入C 59.0g,剧烈搅拌下滴加80wt%水合肼31.3g,滴毕, 130℃温度下反应2~3h后,再升温至210℃反应;
GC检测反应完成,体系降至10~30℃,向其中加入水108ml,分批用108ml正己烷萃取,无水硫酸钠干燥,浓缩,得粗品51.4g,减压蒸馏,收集118℃/5T的馏分,得到D 44.3g,气相质谱纯度98.5%,收率87.1%;
步骤四烯基臭氧化
向反应釜中依次加入二氯甲烷218ml和D 44.3g,降温至-65℃,剧烈搅拌下,向反应釜中通入臭氧,GC检测反应完成,停止通入臭氧,缓慢滴加二甲基硫醚,无剧烈升温后停止滴入,体系温度10~30℃,加入水87ml,分液,二氯甲烷相用无水硫酸钠干燥,浓缩,得液体产品TM41.8g,气相质谱纯度98.5%,收率93.4%。
实施例二
步骤一酰胺化反应
室温下,向反应釜中加入1250ml二氯甲烷和50g 10-十一烯酸,搅拌下滴加129.1g氯化亚砜,滴毕,10℃反应,检测反应完成,浓缩,得到A;
室温下,向反应釜中一次加入344ml二氯甲烷、34.4g N,O-二甲基羟胺盐酸盐,降温至5℃,滴加分子A的二氯甲烷溶液,滴完升至10℃反应;
GC检测反应,反应完成向反应釜中滴加5wt%的稀盐酸400ml,充分搅拌20~60min,分液,水相分批用300ml二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得62.0g B,气相质谱纯度96.3%,收率96.8%;
步骤二格氏反应
向反应釜中依次加入四氢呋喃298ml和B 62.0g,体系降温至25℃,滴加摩尔浓度为1mol/L的异丁基溴化镁263ml,滴加完毕,升至50℃反应;
GC检测反应,反应完成,体系降温至20℃,向其中滴加饱和氯化铵水溶液299ml,分批用300ml二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得55.0g C,气相质谱纯度91.1%,收率85.0%;
步骤三氢化还原
向四口瓶中依次加入二甘醇300ml和氢氧化钾18.8g,升温至70℃,待氢氧化钾溶解后,加入C 55.0g,剧烈搅拌下滴加80wt%水合肼35.0g,滴毕,110℃温度下反应2~3h后,再升温至190℃反应;
GC检测反应完成,体系降至10~30℃,向其中加入水150ml,分批用150ml正己烷萃取,无水硫酸钠干燥,浓缩,得粗品47.2g,减压蒸馏,收集118℃/5T的馏分,得到D 41.4g,气相质谱纯度98.5%,收率86.8%;
步骤四烯基臭氧化
向反应釜中依次加入二氯甲烷331ml和D 41.4 g,降温至-55℃,剧烈搅拌下,向反应釜中通入臭氧,GC检测反应完成,停止通入臭氧,缓慢滴加二甲基硫醚,无剧烈升温后停止滴入,体系温度10~30℃时,加入水82ml,分液,二氯甲烷相用无水硫酸钠干燥,浓缩,得液体产品TM 38.9g,气相质谱纯度99.0%,收率93.5%。
实施例三
步骤一酰胺化反应
室温下,向反应釜中加入2000ml二氯甲烷和100g 10-十一烯酸,搅拌下滴加193.7g氯化亚砜,滴毕升温至30℃反应,检测反应完成,浓缩,得到A;
室温下,向反应釜中一次加入635ml二氯甲烷和63.5g N,O-二甲基羟胺盐酸盐,降温至2℃,滴加分子A的二氯甲烷溶液,滴完升至25℃反应;
GC检测反应,反应完成向反应釜中滴加质量分数12%的稀盐酸500ml,充分搅拌20~60min,分液,水相分批用600ml二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得126.8g B,气相质谱纯度94.7%,收率97.4%;
步骤二格氏反应
向反应釜中依次加入四氢呋喃480ml和B 126.8g,体系降温至10℃,滴加摩尔浓度为1mol/L的异丁基溴化镁528ml,滴加完毕,升至30℃反应;
GC检测反应,反应完成,体系降温至0℃,向其中滴加饱和氯化铵水溶液360ml,分批用650ml二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得112.5g C,气相质谱纯度92.3%,收率87.6%;
步骤三氢化还原
向反应釜中依次加入二甘醇519ml和氢氧化钾36.3g,升温至80℃,待氢氧化钾溶解后,加入C 112.5g,剧烈搅拌下滴加80wt%水合肼66.7g,滴毕,120℃温度下反应2-3h后,再升温至200℃反应;
GC检测反应完成,体系降至10~30℃,向其中加入水260ml,分批用260ml正己烷萃取,无水硫酸钠干燥,浓缩,得粗品100.4g,减压蒸馏,收集118℃/20Pa的馏分,得到D 85.4g,气相质谱纯度99.0%,收率86.8%;
步骤四烯基臭氧化
向反应釜中依次加入二氯甲烷512ml,D 85.4g,降温至-60℃,剧烈搅拌下,向反应釜中通入臭氧,GC检测反应完成,停止通入臭氧,缓慢滴加二甲基硫醚,无剧烈升温后停止滴入,体系温度10~30℃时,加入水127ml,分液,二氯甲烷相用无水硫酸钠干燥,浓缩,得液体产品TM80.0g,气相质谱纯度99.2%,收率93.0%。
实施例四放大实验
步骤一酰胺化反应
室温下,向反应釜中加入6000ml二氯甲烷和377g 10-十一烯酸,搅拌下滴加731g氯化亚砜,滴毕升温至40℃反应,检测反应完成,浓缩,得到A;
室温下,向反应釜中一次加入3000ml二氯甲烷和220g N,O-二甲基羟胺盐酸盐,降温至0℃,滴加A的二氯甲烷溶液,滴毕30℃下反应;
GC检测反应,反应完成向反应釜中滴加质量分数10%的稀盐酸3000ml,充分搅拌20~60min,分液,水相分批用2000ml二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得477.5g B,气相质谱纯度96.3%,收率98.9%;
步骤二格氏反应
向反应釜中依次加入四氢呋喃1500ml和B 477.5g,体系降温至0℃,滴加摩尔浓度为1mol/L的异丁基溴化镁2230ml,滴毕,30℃下反应;
GC检测反应,反应完成,体系降温至0℃,向其中滴加饱和氯化铵水溶液1000ml,分批用2000ml二氯甲烷萃取三次,合并二氯甲烷相,干燥,浓缩,得435.2g C,气相质谱纯度91.8%,收率88.0%;
步骤三氢化还原
向反应釜中依次加入二甘醇1600ml和氢氧化钾150g,升温至80℃,待氢氧化钾溶解后,加入C 435.2g,剧烈搅拌下滴加80wt%水合肼260g,滴毕,120℃温度下反应2~3h后,再升温至200℃反应;
GC检测反应完成,体系降至10~30℃,向其中加入水1000ml,分批用1000ml正己烷萃取,无水硫酸钠干燥,浓缩,得粗品350g,减压蒸馏,收集118℃/5T的馏分,得到D324.4g,气相质谱纯度98.5%,收率85.3%;
步骤四烯基臭氧化
向反应釜中依次加入二氯甲烷2000ml,分子D 324.4g,降温至-60℃,剧烈搅拌下,向反应釜中通入臭氧,GC检测反应完成,停止通入臭氧,缓慢滴加二甲基硫醚,无剧烈升温后停止滴入,体系温度10~30℃时,加入水479ml,分液,二氯甲烷相用无水硫酸钠干燥,浓缩,得液体产品TM 304.8g,气相质谱纯度98.2%,收率92.8%。
- 还没有人留言评论。精彩留言会获得点赞!