用于植物多基因表达载体构建的质粒体系及其应用的制作方法
本发明涉及生物领域,具体涉及一种用于植物多基因表达载体构建的质粒体系及其应用。
背景技术:
植物基因工程技术的迅速发展和广泛应用为植物的遗传改良开拓了广阔的前景。该技术克服了植物有性杂交的限制,可将来源于不同物种甚至人工合成的基因导入植物,从而改良植物性状,培育优质高产作物新品种。在基因克隆、基因功能分析以及基因转化等分子生物学的研究与应用过程中,载体构建是常用的技术手段,采用合适的表达载体和构建方法会使实验效果事半功倍。但是,目前常用于植物基因表达载体构建的质粒,如pBIN19(Bevan,1984)、pBI121(Jefferson,1987)、pIG121-Hm(Ohta et al.,1999)、pMSIsGFP(Kawasaki et al.,1999)等质粒载体,由于其T-DNA领域中限制性内切酶位点有限,目的基因片段难于插入和连接,有时还需要构建多个中间载体,操作比较麻烦,而且这些经典的质粒载体,目前只限于在研究者同行间互相转让,没有商业化,在使用上不尽如人意。为此,一些国际生物大公司,如Invitrogen公司创立了TOPO克隆(Patel,2009)、Gateway重组技术(Chung et al.,2005),ClonTech公司创立了Infusion PCR技术(Berrow et al.,2007)等。TOPO克隆虽然简单,但目的载体范围有限,Gateway和infusion PCR可以不考虑酶切位点,但这些企业开发的技术和载体,主要针对的是动物、微生物的基因表达载体的构建,缺少植物基因表达所必须的启动子、终止子、筛选标记等功能元件,而且费用都比较昂贵(3000-10000元左右),加上由于专利限制,只能应用于研究目的,在应用上受到了限制。另外,目前这些常用的质粒载体和开发的技术,大部分只能构建单个基因表达载体,然而在植物基因改良中,很多性状是由多个基因共同控制的,即要求几个相关基因的协同表达,常常需要构建多个基因表达载体。
技术实现要素:
为解决上述问题,本发明通过对最常用的大肠杆菌质粒载体pUC18和双核农杆菌Ti质粒pBI121进行全方位改造,并插入植物基因表达常用的启动子、终止子、筛选标记等功能元件,同时引入多个克隆酶切位点,建立了一套用于植物单个或多个基因表达载体构建的质粒体系,并提供了该质粒体系的应用。
为实现上述目的,本发明采取的技术方案为:
用于植物多基因表达载体构建的质粒体系,包括pKAFCR0质粒和pKAFCR100质粒;
所述pKAFCR0质粒通过以下步骤构建:
S11、利用限制性内切酶Hind III和EcoR I,对大肠杆菌质粒载体pUC18(Yanisch-Perron et al.,1985)DNA进行双酶切处理;主要目的是切除其多克隆位点MCS(multiple cloning site)的Hind III至EcoR I部分;
S12、同样利用限制性内切酶Hind III和EcoR I,对双核农杆菌Ti质粒pBI121 DNA进行双酶切处理;主要目的是切取其包含植物转基因时最常使用的35S启动子(cauliflower mosaic virus 35S promoter)、GUS(β-glucuronidase)报告基因和NOS终止子(nopaline synthase terminator)的片段。
S13、将双酶切后的pUC18,以及同样双酶切后的pBI121,经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒(如QIAGEN QIAquick Gel Extraction Kit)获得已切除Hind III-EcoR I部分的pUC18和从pBI121切取的35S-GUS-NOS片段;
S14、利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit),将已切除Hind III-EcoR I部分的pUC18和从pBI121切取的35S-GUS-NOS片段进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;
S15、将转化后的大肠杆菌平涂于含有25mg/L青霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒(35S-GUS-NOS片段已插入到pUC18的Hind III和EcoR I位点)的阳性菌落;
S16、将阳性菌落经25mg/L青霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA,该重组质粒命名为pKAFCR1,即在pUC18的Hind III和EcoR I位点上,插入35S-GUS-NOS片段;
S17、利用限制性内切酶Sma I和Sac I,对重组质粒pKAFCR1 DNA进行双酶切处理;主要目的是切除GUS报告基因片段。
S18、将双酶切后的pKAFCR1经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒,获得已切除GUS报告基因片段的pKAFCR1;
S19、人工合成一段DNA,该片段含有限制性内切酶Sma I、Kpn I、Xho I和Sac I位点;利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit)将该DNA片段与切除GUS报告基因的pKAFCR1进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;将转化后的大肠杆菌平涂于含有25mg/L青霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒的阳性菌落;将阳性菌落经25mg/L青霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA;该重组质粒命名为pKAFCR2,即去除了pKAFCR1的GUS报告基因,并在此位置上增加了Kpn I和Xho I酶切位点;
S120、利用限制性内切酶Hind III和Pst I,对重组质粒pKAFCR2 DNA进行双酶切处理;
S121、将双酶切后的pKAFCR2经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒,获得已酶切的pKAFCR2。
S122、人工合成一段DNA,该片段含有限制性内切酶Hind III、Hpa I、Stu I、Sph I和Pst I(Sbf I)位点;利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit)将该DNA片段与已酶切的pKAFCR2进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;将转化后的大肠杆菌平涂于含有25mg/L青霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒的阳性菌落;将阳性菌落经25mg/L青霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA;该重组质粒命名为pKAFCR0,即在35S上游增加了Hpa I、Stu I和Sph I酶切位点;pKAFCR0在35S上游,35S与NOS中间以及在NOS下游均具有多克隆酶切位点;
所述pKAFCR100质粒通过以下步骤构建:
S21、利用限制性内切酶Hind III和EcoR I,对双核农杆菌Ti质粒pBI121DNA进行双酶切处理。主要目的是切除其35S-GUS-NOS片段。
S22、利用限制性内切酶Hind III和EcoR I,对双核农杆菌Ti质粒pBsGFP(Kajiyama et al.,2007)DNA进行双酶切处理;主要目的是切取其35SΩ启动子(35S promoter with an additional omega element translational enhancer)、sGFP(synthetic green-fluorescent protein with S65T mutation)报告基因和NOS终止子的片段。
S23、将双酶切后的pBI121,以及同样双酶切后的pBsGFP,经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒(如QIAGEN QIAquick Gel Extraction Kit),获得已切除35S-GUS-NOS片段的pBI121,以及从pBsGFP切取的35SΩ-sGFP-NOS片段;
S24、利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit),将已切除35S-GUS-NOS片段的pBI121和从pBsGFP切取的35SΩ-sGFP-NOS片段进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;将转化后的大肠杆菌平涂于含有50mg/L卡那霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒的阳性菌落;将阳性菌落经50mg/L卡那霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA,该重组质粒命名为pKAFCR31,即将pBI121的35S-GUS置换成35SΩ-sGFP;
S25、利用限制性内切酶Hind III和Pst I,对重组质粒pKAFCR31 DNA进行双酶切处理;
S26、将双酶切后的质粒pKAFCR31,经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒(如QIAGEN QIAquick Gel Extraction Kit),获得已酶切的pKAFCR31;
S27、人工合成一段DNA,该片段含有限制性内切酶Hind III、Hpa I、Stu I、Sph I和Sbf I位点;利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit),将该DNA片段与已酶切的pKAFCR31进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;将转化后的大肠杆菌平涂于含有50mg/L卡那霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒的阳性菌落;将阳性菌落经50mg/L卡那霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA,该重组质粒命名为pKAFCR100,即在35SΩ启动子上游增加了Hpa I、Stu I和Sph I酶切位点;pKAFCR100具有独立表达的卡那霉素NPT II(neomycin phosphotransferase II)耐性基因和sGFP绿色荧光蛋白报告基因,以及其中间的多克隆酶切位点。
上述的用于植物多基因表达载体构建的质粒体系可用于植物单个或多个基因表达载体的构建。
本发明具有以下有益效果:
1、改造形成的重组质粒pKAFCR0具有植物基因表达最常使用的35S启动子和NOS终止子,利用PCR等方法克隆得到的目的基因片段可以多种方式很方便地连接到pKAFCR0的35S与NOS中间。
2、改造形成的重组质粒pKAFCR100具有独立表达的卡那霉素NPT II耐性基因和sGFP绿色荧光蛋白报告基因,以及其中间的多克隆酶切位点。
3、利用pKAFCR0与pKAFCR100质粒的配套使用,可以方便地构建植物单个或多个基因表达载体。由此方法构建的基因表达载体,目的基因的上游和下游分别连接有植物体内基因表达所必须的启动子和终止子,使插入的目的基因能够在植物体内稳定表达;同时具有独立表达的卡那霉素NPT II耐性基因和sGFP绿色荧光蛋白报告基因等筛选标记,可用于转基因过程细胞的选拔和观察,有效提高转基因效率。
4、目的基因上游和下游的35S启动子和NOS终止子、NPT II抗生素耐性基因和sGFP绿色荧光蛋白报告基因前后均有酶切位点,可以根据实验需要选择更合适的基因元件对其进行更换。
附图说明
图1为本发明实施例中pKAFCR0质粒多克隆位点和pKAFCR100质粒T-DNA示意图。
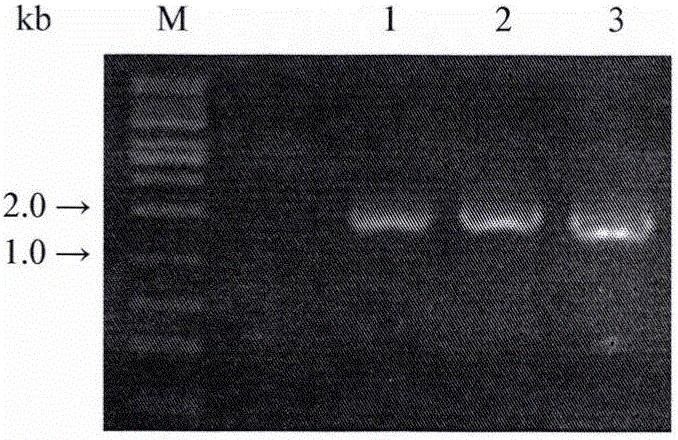
图2为本发明实施例中SrUGT76G1、SrUGT85C2和SrUGT91D2m基因全长PCR扩增。图中:M:DNA Marker;1:SrUGT76G1;2:SrUGT85C2;3:SrUGT91D2m。
图3为本发明实施例中SrUGT76G1、SrUGT85C2和SrUGT91D2m目的基因单独连接至pKAFCR0质粒后的菌落PCR鉴定。图中:M:DNA Marker;1-4:SrUGT76G1(4为阴性);5-8:SrUGT85C2(6为阴性);9-12:SrUGT91D2m。
图4为本发明实施例中SrUGT76G1、SrUGT85C2和SrUGT91D2m基因组合PCR扩增。图中:M:DNA Marker;1-3:SrUGT76G1;4-6:SrUGT91D2m;7-9:SrUGT85C2。
图5为本发明实施例中SrUGT76G1、SrUGT85C2和SrUGT91D2m目的基因组合单独连接至pKAFCR100质粒后的菌落PCR鉴定。图中:M:DNA Marker;1-3:SrUGT76G1;4-6:SrUGT85C2;7-9:SrUGT91D2m(7为阴性)。
图6为本发明实施例中SrUGT76G1、SrUGT85C2和SrUGT91D2m目的基因组合单独构建至pKAFCR100后的重组质粒示意图。图中:上:pNULPGE01101(pKAFCR100+SrUGT76G1基因组合);中:pNULPGE01102(pKAFCR100+SrUGT85C2基因组合);下:pNULPGE01103(pKAFCR100+SrUGT91D2m基因组合)。
图7为本发明实施例中SrUGT76G1、SrUGT85C2和SrUGT91D2m 3个目的基因组合全部连接至pKAFCR100质粒后的菌落PCR鉴定。图中:M:DNA Marker;1-5:SrUGT76G1;6-10:SrUGT85C2;11-15:SrUGT91D2m。
图8为本发明实施例中SrUGT76G1、SrUGT85C2和SrUGT91D2m3个目的基因组合全部连接至pKAFCR100后的重组质粒pNULPGE01105示意图。
具体实施方式
为了使本发明的目的及优点更加清楚明白,以下结合实施例对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
用于植物多基因表达载体构建的质粒体系,包括pKAFCR0质粒和pKAFCR100质粒;
所述pKAFCR0质粒通过以下步骤构建:
S11、利用限制性内切酶Hind III和EcoR I,对大肠杆菌质粒载体pUC18(Yanisch-Perron et al.,1985)DNA进行双酶切处理;主要目的是切除其多克隆位点MCS(multiple cloning site)的Hind III至EcoR I部分;
S12、同样利用限制性内切酶Hind III和EcoR I,对双核农杆菌Ti质粒pBI121 DNA进行双酶切处理;主要目的是切取其包含植物转基因时最常使用的35S启动子(cauliflower mosaic virus 35S promoter)、GUS(β-glucuronidase)报告基因和NOS终止子(nopaline synthase terminator)的片段。
S13、将双酶切后的pUC18,以及同样双酶切后的pBI121,经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒(如QIAGEN QIAquick Gel Extraction Kit)获得已切除Hind III-EcoR I部分的pUC18和从pBI121切取的35S-GUS-NOS片段;
S14、利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit),将已切除Hind III-EcoR I部分的pUC18和从pBI121切取的35S-GUS-NOS片段进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;
S15、将转化后的大肠杆菌平涂于含有25mg/L青霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒(35S-GUS-NOS片段已插入到pUC18的Hind III和EcoR I位点)的阳性菌落;
S16、将阳性菌落经25mg/L青霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA,该重组质粒命名为pKAFCR1,即在pUC18的Hind III和EcoR I位点上,插入35S-GUS-NOS片段;
S17、利用限制性内切酶Sma I和Sac I,对重组质粒pKAFCR1 DNA进行双酶切处理;主要目的是切除GUS报告基因片段。
S18、将双酶切后的pKAFCR1经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒,获得已切除GUS报告基因片段的pKAFCR1;
S19、人工合成一段DNA,该片段含有限制性内切酶Sma I、Kpn I、Xho I和Sac I位点;利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit)将该DNA片段与切除GUS报告基因的pKAFCR1进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;将转化后的大肠杆菌平涂于含有25mg/L青霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒的阳性菌落;将阳性菌落经25mg/L青霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA;该重组质粒命名为pKAFCR2,即去除了pKAFCR1的GUS报告基因,并在此位置上增加了Kpn I和Xho I酶切位点;
S120、利用限制性内切酶Hind III和Pst I,对重组质粒pKAFCR2 DNA进行双酶切处理;
S121、将双酶切后的pKAFCR2经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒,获得已酶切的pKAFCR2。
S122、人工合成一段DNA,该片段含有限制性内切酶Hind III、Hpa I、Stu I、Sph I和Pst I(Sbf I)位点;利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit)将该DNA片段与已酶切的pKAFCR2进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;将转化后的大肠杆菌平涂于含有25mg/L青霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒的阳性菌落;将阳性菌落经25mg/L青霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA;该重组质粒命名为pKAFCR0,即在35S上游增加了Hpa I、Stu I和Sph I酶切位点;pKAFCR0在35S上游,35S与NOS中间以及在NOS下游均具有多克隆酶切位点;如图1所示。
所述pKAFCR100质粒通过以下步骤构建:
S21、利用限制性内切酶Hind III和EcoR I,对双核农杆菌Ti质粒pBI121 DNA进行双酶切处理。主要目的是切除其35S-GUS-NOS片段。
S22、利用限制性内切酶Hind III和EcoR I,对双核农杆菌Ti质粒pBsGFP(Kajiyama et al.,2007)DNA进行双酶切处理;主要目的是切取其35SΩ启动子(35S promoter with an additional omega element translational enhancer)、sGFP(synthetic green-fluorescent protein with S65T mutation)报告基因和NOS终止子的片段。
S23、将双酶切后的pBI121,以及同样双酶切后的pBsGFP,经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒(如QIAGEN QIAquick Gel Extraction Kit),获得已切除35S-GUS-NOS片段的pBI121,以及从pBsGFP切取的35SΩ-sGFP-NOS片段;
S24、利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit),将已切除35S-GUS-NOS片段的pBI121和从pBsGFP切取的35SΩ-sGFP-NOS片段进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;将转化后的大肠杆菌平涂于含有50mg/L卡那霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒的阳性菌落;将阳性菌落经50mg/L卡那霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA,该重组质粒命名为pKAFCR31,即将pBI121的35S-GUS置换成35SΩ-sGFP;
S25、利用限制性内切酶Hind III和Pst I,对重组质粒pKAFCR31 DNA进行双酶切处理;
S26、将双酶切后的质粒pKAFCR31,经1%的琼脂糖凝胶电泳分离后,切取目的条带,通过凝胶回收方法或凝胶回收试剂盒(如QIAGEN QIAquick Gel Extraction Kit),获得已酶切的pKAFCR31;
S27、人工合成一段DNA,该片段含有限制性内切酶Hind III、Hpa I、Stu I、Sph I和Sbf I位点;利用T4-DNA连接酶或质粒连接用试剂盒(如TOYOBO Ligation High Kit),将该DNA片段与已酶切的pKAFCR31进行连接;连接后的重组质粒,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中;将转化后的大肠杆菌平涂于含有50mg/L卡那霉素的LB固体培养基上培养,挑取单独菌落进行菌落PCR鉴定,筛选含有重组质粒的阳性菌落;将阳性菌落经50mg/L卡那霉素的LB液体培养基扩繁后,利用常规的质粒提取方法或质粒提取试剂盒(如QIAGEN Qiaprep Spin Miniprep Kit)提取重组质粒DNA,该重组质粒命名为pKAFCR100,即在35SΩ启动子上游增加了Hpa I、Stu I和Sph I酶切位点;pKAFCR100具有独立表达的卡那霉素NPT II(neomycin phosphotransferase II)耐性基因和sGFP绿色荧光蛋白报告基因,以及其中间的多克隆酶切位点,如图1所示。
上述的用于植物多基因表达载体构建的质粒体系可用于植物单个或多个基因表达载体的构建,具体的有以下方法:
1、目的基因的上游和下游分别加上启动子和终止子(目的基因与pKAFCR0质粒的连接)
因为pKAFCR0在35S与NOS中间具有多克隆酶切位点,利用PCR等方法克隆得到的目的基因片段可以很方便地连接到pKAFCR0的35S与NOS中间,在目的基因的上游和下游分别加上植物体内基因表达所必须的启动子和终止子:
(1)目的基因PCR扩增产物的5’、3’两端酶切后为粘性末端的连接
目的基因在PCR扩增时,通过正反向引物的5’端引入与pKAFCR0在35S-NOS中间相同的粘性末端酶切位点(Xba I、BamH I、Kpn I、Xho I和Sac I的其中一种),这样扩增后的PCR产物经酶切后5’、3’两端均为粘性末端,可以与经同样酶切后的pKAFCR0进行连接。如果两末端是相同的酶切位点,连接后还需要进行插入片段的方向判断。
(2)目的基因PCR扩增产物的5’、3’两端酶切后其中一段为粘性末端,另一端为平滑末端的连接
目的基因在PCR扩增时,通过正向引物,或者反向引物的5’端引入与pKAFCR0在35S-NOS中间相同的粘性末端酶切位点(正向引物引入Xba I和BamH I的其中一种,或者反向引物引入Kpn I、Xho I和Sac I的其中一种),而另一向引物的5’端引入任何一种平滑末端酶切位点,这样扩增后的PCR产物经酶切后其中一端为粘性末端,另一端为平滑末端,可以与一端经同样粘性末端酶切,另一端经平滑末端酶切(Xma I或Sma I的其中一种)后的pKAFCR0进行连接。
(3)目的基因PCR扩增产物的5’、3’两端酶切后均为平滑末端的连接
目的基因在PCR扩增时,通过正向引物和反向引物的5’端引入任何一种平滑末端酶切位点,这样扩增后的PCR产物经酶切后两末端均为平滑末端,可以与经平滑末端酶切(Xma I或Sma I的其中一种)后的pKAFCR0进行连接。连接后需要进行插入片段的方向判断。
(4)目的基因PCR扩增产物的5’、3’两端均为平滑末端的连接
利用有些DNA聚合酶(如PHUSION高保真DNA聚合酶),在PCR扩增后能够产生平滑末端产物的特点,可以直接与经平滑末端酶切(Xma I或Sma I的其中一种)后的pKAFCR0进行连接。连接后需要进行插入片段的方向判断。
目的基因在PCR扩增时,也可以通过正向引物,或者反向引物的5’端引入与pKAFCR0在35S-NOS中间相同的粘性末端酶切位点(正向引物引入Xba I和BamH I的其中一种,或者反向引物引入Kpn I、Xho I和Sac I的其中一种),而另一向引物的5’端不引入酶切位点,这样PCR扩增后虽然产生两端均为平滑末端的产物,但其中一端酶切后为粘性末端,而另一端不酶切仍然保持为平滑末端,可以与一端经同样粘性末端酶切,另一端经平滑末端酶切(Xma I或SmaI的其中一种)后的pKAFCR0进行连接。
(5)目的基因与pKAFCR0质粒连接后的测序检验
目的基因与pKAFCR0质粒的连接后,可以利用pKAFCR0的特异引物,如CR0 MCS insert 35S-S(5’-CAC AAT CCC ACT ATC CTT CGC A-3’)和CR0 MCS insert NOS-A(5’-TCC ACT CTA ATC ATA AAA ACC CAT CTC-3’),或者利用pUC18的常用引物,如M13-F(5’-GTA AAA CGA CGG CCA GT-3和M13-R(5’-GGA AAC AGC TAT GAC CAT G-3’)进行测序,进行测序,检验插入的目的基因是否正确。
2、植物单个基因表达载体的构建(单个目的基因组合与pKAFCR100质粒的连接)
目的基因的上游和下游分别加上启动子和终止子的片段称为基因组合(Cassette)。上述目的基因与pKAFCR0质粒连接后,利用pKAFCR0在35S上游和NOS下游具有的多克隆酶切位点(35S上游为Pvu II、Hind III、Hpa I、Stu I、Sph I和Pst I(Sbf I),NOS下游为EcoR I和Pvu II),经酶切后获得目的基因组合片段。该基因组合片段与同样酶切后的pKAFCR100质粒进行连接,最后获得可以在植物体内进行基因表达的载体。
由此方法构建的基因表达载体,目的基因的上游和下游分别连接有植物体内基因表达所必须的启动子和终止子,同时具有独立表达的卡那霉素NPT II耐性基因和sGFP绿色荧光蛋白报告基因,前者可以利用添加有卡那霉素的培养基对转基因植物细胞进行选拔,后者可以利用荧光显微镜对转基因植物细胞进行判断和观察。
另外,目的基因上游和下游的35S启动子和NOS终止子、NPT II抗生素耐性基因和sGFP绿色荧光蛋白报告基因前后均有酶切位点,也可以根据实验需要选择更合适的基因元件对其进行更换(图1)。
3、植物多个基因表达载体的构建(多个目的基因组合与pKAFCR100质粒的连接)
第一个目的基因与pKAFCR0质粒连接后,利用pKAFCR0在35S上游的粘性末端酶切位点(Hind III),和NOS下游的平滑末端酶切位点(Pvu II)进行酶切;或者利用NOS下游的粘性末端酶切位点(EcoR I)酶切后,再进行平滑化处理,获得一个5’端为粘性末端、3’端为平滑末端的目的基因组合片段。而pKAFCR100质粒则进行同样的粘性末端酶切(Hind III),和不同的平滑末端酶切(Hpa I或Stu II),这样酶切后的目的基因组合片段与酶切后的pKAFCR100质粒进行连接后,形成的新质粒在连接处的5’端仍然保留原有的粘性末端酶切位点(Hind III),而3’端因为是不同的平滑末端酶切,连接后平滑末端酶切位点消失。但是由于插入的目的基因组合在35S的上游具有同样的粘性末端酶切位点(Hind III),和平滑末端酶切位点(Hpa I或Stu II),与pKAFCR0质粒连接后的第二个目的基因,同样采用上述酶切方法,获得一个5’端为粘性末端、3’端为平滑末端的目的基因组合片段,连接到已有第一个目的基因组合的质粒上。
这样巧妙利用pKAFCR0与pKAFCR100质粒具有部分相同的酶切位点,两个质粒的配套使用,可以把多个目的基因组合片段连接到pKAFCR100质粒,构建成植物多基因表达载体。
如果目的基因组合片段与pKAFCR100质粒没有匹配的酶切位点,也可以再次对目的基因组合片段进行PCR扩增。利用上述在PCR扩增时,通过正向引物在5’端引入与pKAFCR100质粒具有相同的粘性末端酶切位点(Cla I或Ase I),而利用能够产生平滑末端产物的DNA聚合酶(如PHUSION高保真DNA聚合酶)在3’端形成平滑末端,这样也可以不断地把多个目的基因组合片段连接到pKAFCR100质粒。
实施例
甜叶菊葡萄糖基转移酶SrUGT76G1、SrUGT85C2和SrUGT91D2m基因的克隆与载体构建:
1、甜叶菊叶片总RNA的提取与cDNA第一条链的合成
以甜叶菊品系中山二号的叶片为实验材料,用量为80-100mg。利用RNAprep Pure多糖多酚植物总RNA提取试剂盒(天根)提取总RNA。提取的总RNA经NanoDrop 2000(Thermo)测定浓度后,用RNase-free ddH2O稀释成100ng/μL溶液,利用TransScript Fly First-Strand cDNA Synthesis SuperMix 试剂盒(全式金)进行cDNA第一条链的合成。具体操作步骤按试剂盒的使用说明。
2、目的基因全长PCR扩增和产物提纯
(1)引物设计:参照NCBI等数据库公开的甜叶菊SrUGT76G1(AY345974)、SrUGT85C2(AY345978)和SrUGT91D2m(EU751291)基因的序类,设计各基因PCR扩增引物,引物的5’端引入与pKAFCR0在35S-NOS中间相同的粘性末端酶切位点:
SrUGT76G1、SrUGT85C2和SrUGT91D2m目的基因全长PCR扩增引物
(2)PCR扩增:以上述合成的cDNA(50ng/μL)为模板,使用高保真Phusion High-Fidelity DNA Polymerase PCR(Thermo)试剂盒对上述基因的全长进行扩增(图2),PCR反应条件为98℃ 30s,94℃ 10s 60℃ 30s 72℃ 45s 30cycles,72℃ 7min,4℃。具体操作步骤按试剂盒的使用说明。
(3)PCR产物提纯:将上述PCR产物2μL电泳检测后,利用QIAquick PCR Purification Kit试剂盒(QIAGEN)提纯PCR产物,具体操作步骤按试剂盒的使用说明。
3、pKAFCR0和pKAFCR100质粒DNA的提取
利用Qiaprep Spin Miniprep Kit试剂盒(QIAGEN),从大肠杆菌DH5α提取pKAFCR0和pKAFCR100质粒DNA,具体操作步骤按试剂盒的使用说明。利用NaroDrop 2000测定其浓度。
4、单个目的基因与pKAFCR0质粒的连接
(1)双酶切处理:使用限制性内切酶Kpn I和Sac I分别对经PCR扩增、提纯所得到的SrUGT85C2和SrUGT91D2m目的基因全长以及pKAFCR0质粒进行双酶切处理,而使用限制性内切酶BamH I和Xho I对经PCR扩增得到的SrUGT76G1目的基因全长和pKAFCR0质粒进行双酶切处理。双酶切处理的反应条件为37℃ 4h,灭活60℃ 5min。
(2)目的片段回收:将经双酶切处理后的反应产物进行电泳,检测目的基因片段以及质粒的酶切效果,然后用QIAquick Gel Extraction Kit试剂盒(QIAGEN)对目的片段进行胶回收,具体操作步骤按试剂盒的使用说明。
(3)连接反应:将相同酶切后经胶回收得到的目的基因片段分别与pKAFCR0进行连接,分别按目的基因片段125fmol、质粒25fmol的比例,经过65℃ 5min前处理以消除DNA形成二级结构后,加入半量体积的Ligation High连接酶(TOYOBO),在16℃ 12h条件下进行连接反应。
(4)大肠杆菌基因转化:取适量(<10μL)已连接好的反应产物,利用热激发转化到大肠杆菌DH5α菌株的感受态细胞中。
(5)菌落PCR鉴定:将质粒转化后的大肠杆菌液(100μL)涂在固体LB抗生素培养基(青霉素25mg/L)上培养一晚,挑取单个菌落,利用上述引物进行菌落PCR鉴定(图3)。将PCR阳性菌落置于液体LB抗生素培养基(青霉素25mg/L)培养一晚,再次PCR鉴后,保存阳性菌液。
(6)目的基因插入测序:将保存好的阳性菌液扩繁后,利用Qiaprep Spin Miniprep Kit试剂盒,提取构建后的质粒DNA送至invitrogen公司进行测序验证。测序用引物为CR0 MCS insert 35S-S和CR0 MCS insert NOS-A。
目的基因成功构建至pKAFCR0的重组质粒分别命名为:
pNULPGE01001:pKAFCR0+SrUGT76G1
pNULPGE01002:pKAFCR0+SrUGT85C2
pNULPGE01003:pKAFCR0+SrUGT91D2m
5、构建单基因表达载体
由于3个目的基因组合(目的基因构建至pKAFCR0后,其上游和下游分别加上启动子和终止子的片段)都没有Ase I酶切位点,以目的基因构建至pKAFCR0的重组质粒DNA为模板,利用PCR扩增方法,通过正向引物在5’端引入Ase I粘性末端酶切位点,而利用能够产生平滑末端产物的DNA聚合酶(如PHUSION高保真DNA聚合酶)在3’端形成平滑末端,这样的PCR扩增产物,经Ase I酶切处理后,可以与经Ase I和Stu I酶切处理后的pKAFCR100进行连接,构建单基因表达载体。连接处Ase I粘性末端酶切位点仍然保留,而Stu I平滑末端酶切位点消失。
(1)重组质粒DNA提取:从大肠杆菌DH5α中分别提取各目的基因构建至pKAFCR0的重组质粒pNULPGE01001、pNULPGE01002和pNULPGE01003。利用NaroDrop 2000测定其浓度。
(2)PCR扩增和产物提纯:目的基因组合的PCR扩增引物设计如下,引物的5’端引入Ase I粘性末端酶切位点:
目的基因组合的PCR扩增引物
重组质粒pNULPGE01001、pNULPGE01002、pNULPGE01003 DNA稀释成10ng/μL后,分别以此为模板,使用高保真Phusion High-Fidelity DNA Polymerase PCR试剂盒进行PCR扩增(图4),PCR反应体系和条件同上。将上述PCR产物2μL电泳检测后,利用QIAquick PCR Purification Kit试剂盒提纯PCR产物。
(3)目的基因组合PCR扩增产物和pKAFCR100双酶切处理:将提纯的目的基因组合的PCR扩增产物用Ase I进行酶切,pKAFCR100用Ase I和Stu I进行双酶切。酶切反应条件为:37℃ 4h,90℃ 5min。
(4)目的片段回收:将上述经酶切处理后的反应产物进行电泳检测,然后用QIAquick Gel Extraction Kit试剂盒对目的片段进行胶回收。
(5)连接反应:将胶回收得到的目的基因组合与胶回收得到的pKAFCR100,按上述方法进行连接、大肠杆菌转化(培养基的抗生素改为卡那霉素50mg/L)以及菌落PCR鉴定后(图5),保存阳性菌落。目的基因组合成功构建至pKAFCR100的重组质粒(图6)分别命名为:
pNULPGE01101:pKAFCR100+SrUGT76G1基因组合;
pNULPGE01102:pKAFCR100+SrUGT85C2基因组合;
pNULPGE01103:pKAFCR100+SrUGT91D2m基因组合。
6、构建两个基因表达载体
构建单基因表达载体时,目的基因组合与pKAFCR100的连接处Ase I粘性末端酶切位点仍然保留,而Stu I平滑末端酶切位点消失。但是由于插入的目的基因组合在35S的上游具有同样的粘性末端酶切位点Ase I和Stu I,第二个目的基因组合可以连接到已有第一个目的基因组合的质粒上。
(1)重组质粒DNA提取:从大肠杆菌DH5α中提取SrUGT85C2目的基因组合构建至pKAFCR100的重组质粒pNULPGE01102。利用NaroDrop 2000测定其浓度。
(2)pNULPGE01102双酶切处理:用Ase I和Stu I进行双酶切。酶切反应条件为:37℃ 4h,90℃ 5min。
(3)目的片段回收:将上述经酶切处理后的反应产物进行电泳检测,然后用QIAquick Gel Extraction Kit试剂盒对目的片段进行胶回收。
(4)连接反应:将5(4)胶回收得到的SrUGT91D2m目的基因组合与胶回收得到的pNULPGE01102,按上述方法进行连接、大肠杆菌转化(培养基的抗生素改为卡那霉素50mg/L)以及菌落PCR鉴定后,保存阳性菌落。两个目的基因组合成功构建至pKAFCR100的重组质粒命名为:
pNULPGE01104:pKAFCR100+SrUGT91D2m基因组合+SrUGT85C2基因组合。
7、构建三个基因表达载体
(1)重组质粒DNA提取:从大肠杆菌DH5α中提取重组质粒pNULPGE01104。利用NaroDrop 2000测定其浓度。
(2)pNULPGE01104双酶切处理:用Ase I和Stu I进行双酶切。酶切反应条件为:37℃ 4h,90℃ 5min。
(3)目的片段回收:将上述经酶切处理后的反应产物进行电泳检测,然后用QIAquick Gel Extraction Kit试剂盒对目的片段进行胶回收。
(4)连接反应:将5(4)胶回收得到的SrUGT76G1目的基因组合与胶回收得到的pNULPGE01104,按上述方法进行连接、大肠杆菌转化(培养基的抗生素改为卡那霉素50mg/L)以及菌落PCR鉴定后(图7),保存阳性菌落。三个目的基因组合成功构建至pKAFCR100的重组质粒(图8)命名为:
pNULPGE01105:pKAFCR100+SrUGT76G1目的组合+SrUGT91D2m基因组合+SrUGT85C2基因组合。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以作出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!