一类2,3‑内酰胺环稠合喹唑啉‑4(3H)‑酮衍生物及其制备方法和应用与流程
本发明属于药物制备技术领域,涉及药物化学基先导化合物的研发制备,具体涉及一类2,3-内酰胺环稠合喹唑啉-4(3H)-酮衍生物及其制备方法和应用。
背景技术:
作为一种药物化学的优势骨架,喹唑啉构成了诸多具有广泛生物活性的化合物,如抗炎、抑菌、抗结核、抗糖尿病、抗HIV和抗肿瘤等活性。很多喹唑啉类化合物对多种抗肿瘤药物靶标,如EGFR,PARP,MPS1,JAK2,CHK-2及Pinl等,均显示明显的抑制活性。新近上市的作为酪氨酸激酶抑制剂的抗肿瘤药物吉非替尼(Gefitinib)、厄洛替尼(Erlotinib)和拉帕替尼(Lapatinib)都含有喹唑啉的杂环结构特征,结构如下:
不仅如此,稠合的喹唑啉-4-酮还广泛存在于各种活性天然产物的结构之中,同时也是制备大量具有显著生物活性的生物碱类化合物的重要建筑模块。大约有150中天然的生物碱类化合物中含有稠合的喹唑啉-4-酮结构片段,其中有很多稠环结构具有抗肿瘤活性,如:色胺酮(Tryptanthrine)、达尼喹酮(Batracyclin)、鸭嘴花碱酮(Vasicinone)、骆驼宁碱A(Luotonin A)、吴茱萸次碱(Ruteacarpine)和吴茱萸碱(Evodiamine)等。这些化合物具有类似的三环稠和喹唑啉杂环体系,包括A,B和C环作为他们共有的主要结构骨架(2,3-稠合的(3H)-喹唑啉-4-酮类化合物,结构如下:
上述稠合喹唑啉-4-酮类化合物分别具有各种各样的抗肿瘤活性,下文将分别阐述之。
色胺酮(Tryptanthrine)为吲哚喹唑啉类生物碱,主要存在于马蓝、蓼蓝、菘蓝等产蓝植物中,是中药青黛和大青叶的有效成分。作为一种新型芳香化合物受体的拮抗剂,色胺酮及其衍生物具有抗炎、抗菌、预防癌症、抗肿瘤(如抑制耐药性的癌细胞和对乳腺癌细胞及逆转阿霉素的耐药性)、抗真菌、抗疟、抗寄生虫、抗白血病、等广泛生物活性。
达尼喹酮(Daniquidone,Batracyclin,nsc-320846)是一种具有细胞毒性的抗癌试剂,不仅在小鼠体内显示强烈抗实体肿瘤活性,且对阿霉素、顺铂和甲氨蝶呤产生耐药性的小鼠白血病P-388细胞及结肠癌38细胞均有抑制效果。该化合物可作为拓扑异构酶1和2的双重抑制剂,稳定DNA/酶的二联复合物,诱发大鼠肝细胞的非增殖细胞计划外DNA合成,显示出对HT29结肠癌细胞株生长抑制作用(GI50=10μM)。
鸭嘴花碱酮(Vasicinone)是一种在爵床科植物鸭嘴花叶中大量存在的生物碱,在传统印度医学中广为人知。早期发现它是具有扩张支气管的生物活性。近来研究报道其具有抗炎、抗菌、抗哮喘、保肝和中等程度的抗癌活性。鸭嘴花碱酮扩环衍生物RLX对各种癌细胞株可诱发意义深远的抗增生活性,如含有HCT-116细胞株的结肠癌细胞对RLX-介导的增生抑制作用最为敏感。RLX可作用于PI3K/Akt/FoxO3a信号传导通路,并可作为放大PI3K(PI3KCA)并逆转肿瘤发展,RLX抗肿瘤活性另一个可能机制是通过引发人类结肠癌细胞株HCT-116NFκB细胞特异性的凋亡。
蒺藜科植物骆驼蒿在中国传统中长期用于治疗风湿,脓肿等炎症性疾病,与其含有抗肿瘤生物碱有关。从骆驼蒿中分离出来的骆驼宁A是一种天然吡咯喹唑啉酮生物碱,已经被证实具有抗肿瘤活性,对肺癌、宫颈癌、乳腺癌及阿霉素耐药的乳腺癌细胞株均有抑制作用。骆驼宁A对白血病细胞株P-388抑制作用(IC50=1.8μg/mL)引发了人们的浓厚研究兴趣。进而研究了骆驼宁碱产生细胞毒性的作用机制,发现它可作为一种拓扑异构酶I-DNA抑制剂,通过与拓扑异构酶I-DNA的共价二元配合物结合来抑制完整细胞中拓扑异构酶I依赖性细胞毒性(IC50=5.7–12.6μg/mL)。抗癌生物碱骆驼宁碱A的生物活性作用机理可与喜树碱媲美,都属于拓扑异构酶I抑制剂,二者的化学结构骨架都由相同的ABC环片断构成,只有D环存在轻微不同(喜树碱为吡啶酮,骆驼宁A中为嘧啶酮),E环存在很大不同。尽管骆驼宁碱类化合物的抗肿瘤活性及细胞选择性不及喜树碱类强,但结构上的相似性足以用骆驼宁碱类化合物作为先导化合物去合成更加强效的潜在抗肿瘤药物。然而,骆驼宁A与拓扑异构酶-I-DNA2分子复合物有可能异于一些喜树碱的结合程度和结合模式。骆驼宁A没有喜树碱敏感的羟基内酯结构和手性中心,所以,就化学稳定性和易合成性上更具优势。另一方面,未改性的骆驼宁A的细胞毒素活性比喜树碱低,激励了许多改变骆驼宁A结构方向的研究以鉴定更多高效衍生物。其中环A的取代被认为是系统变化更具前景的方向。研究发现一些生物碱A环取代衍生物显示出比先导化合物更高的活性。
吴茱萸次碱(Rutaecarpine)具有多种生物活性如利尿、发汗、舒张血管、抗缺氧、改善脑功能、抗血小板聚集以及抗伤害感受等,其中抗伤害感受被认为由抗炎作用介导,但抗炎作用机理尚不明确。吴茱萸次碱对脂多糖(LPS)刺激巨噬细胞产生前列腺素(PG)的作用。吴茱萸次碱还具有抗血栓抑制血小板作用、血管松弛作用、肛门括约肌松弛作用和COX2抑制作用,吴茱萸次碱对小鼠肝肾细胞色素P450依赖的单加氧酶有诱导作用,对人和小鼠肝微粒体细胞色素P4501A有选择性抑制作用,对K+通道有阻滞作用。吴茱萸碱(Evodiamine)具有抗肿瘤生长、收缩支气管、抗伤害感受、舒张血管、抗NO产生等作用。其发现其具有体外抗肿瘤细胞迁移的活性(IC50为1.25ug/mL)。可诱导人宫颈癌Hela细胞发生Capase蛋白酶依赖性的凋亡,在肿瘤转移治疗中具有广阔前景。另外,吴茱萸碱(Evodiamine)和吴茱萸次碱(Rutaecarpine)对三重阴性,包括雌激素受体(ER),孕激素受体(PR)和人类雌激素受体2(HER2)低表达,乳腺癌细胞株MDA-MB-231细胞株的半数抑菌浓度(IC50)分别为33.4mM和73.4mM。而作为雌激素受体α的变种,雌激素受体亚型α36对内分泌耐药和雌激素的非基因活性具有显著功能。鉴于在MDA-MB-231细胞株的细胞膜和细胞质中雌激素受体亚型α36都是高表达的,推测该类化合物也有可能是通过与雌激素受体亚型(ER-α36)相互作用而产生抗肿瘤活性的。
上述天然产物的结构都是2,3-稠合的(3H)-喹唑啉-4-酮类化合物,而人工合成的稠合喹唑啉-4-酮类化合物的研究主要集中在对其芳香性A环的稠合研究。如1996年Rewcastle课题组报道了间溴苯氨基取代的稠环类喹唑啉化合物,对EGFR具有极强的抑制作用喹唑啉并咪唑化合物1(结构式如下)对EGFR的抑制活性IC50值在0.008nM水平。该课题组重点对杂环的位置和类型进行了研究。研究发现,当咪唑与喹唑啉5,6-位稠合后化合物活性显著降低(IC50=29nM);当与7,8-位稠合时,化合物活性又降低10倍(IC50=272nM)。表明直线型稠合结构对化合物的活性有利。对咪唑N-1为甲基化得到化合物2(IC50=0.01nM)(结构式如下),活性略有降低;对喹唑啉C-5位甲基化,化合物保持了较强的活性(IC50=0.025nM)。表明该处对疏水性基团有一定耐受性。将咪唑替换为其它杂环基团,如噻唑、1,2,3-三唑、吡唑、吡嗪等基团,化合物的活性降低;当替换为苯环时,化合物3的活性提高(结构式如下),IC50=0.003nM,但溶解度降低。化合物1具有好选择性,对表皮瘤细胞A431中EGFR抑制活性IC50值为46nM。后续研究表明该类结构具有多种生物活性,如对抗糖尿病靶标果糖-1,6-二磷酸酶(F16BP)的抑制活性为4μM。除此之外,吡唑[1,5-c]并喹唑啉类衍生物还被用作Gly/NMDA受体和有效的氨基酸受体拮抗剂;吡唑[1,5-c]并喹唑啉类衍生物作为红藻氨酸受体拮抗剂;三唑并喹唑啉类衍生物可作为氨基丙酸类受体拮抗剂,用于治疗帕金森综合征。
综上所述,简单喹唑啉环类化合物可作用于酪氨酸激酶而具有抗肿瘤活性,而多环稠合喹唑啉类化合物作用于拓扑异构酶而表现出抗肿瘤活性,但是介于二者之间A环稠环喹唑啉类化合物又显示出较好的抗肿瘤活性,因此可通过将多环稠合喹唑啉类化合物进行结构简化获得容易合成的化合物,而有关喹唑啉的B环进行稠合的三环类化合物的研究依然比较薄弱,由于线性A环稠合结构显示较好的抗癌活性,法国科学家曾试图通过特殊试剂Appel盐(4,5-二氯-1,2,3-二硫代鎓盐氯化物)合成这种特殊酮基取代的三环喹唑啉酮类衍生物,原本设计的三环化合物羰基化合物4却没有得到,却得到了亚氨基取代产物5,如下式:
上述稠环结构多半是在喹唑啉的A环上并合一个杂环,获得了一定研究进展,而对其B环的并合研究目前研究的不是很多。鉴于上述研究背景,为寻找新颖的简单结构母核的抗肿瘤先导化合物,综合考虑这些具有抗肿瘤活性的天然产物的共同结构母核,从其共同的多环平面结构出发,设计简化的平面三环结构为一种结构简单三环骨架则可兼容这些化合物的共同特征母核。
技术实现要素:
本发明的目的在于提供一类2,3-内酰胺环稠合喹唑啉-4(3H)-酮衍生物及其制备方法和应用。
本发明是通过以下技术方案来实现:
本发明公开了一类2,3-内酰胺环稠合喹唑啉-4(3H)-酮衍生物,其结构式如下:
其中,R为H,Br或NO2;n=1或2或3。
本发明还公开了制备上述2,3-内酰胺环稠合喹唑啉-4(3H)-酮衍生物的制备方法,包括以下步骤:
1)以邻氨基苯甲酰胺为原料,与草酸二乙酯稠合构建A、B环,制得中间体8;
2)将中间体8与二溴代烷烃,在干燥的DMF中以碳酸钾作为缚酸剂进行烷基化反应,制得中间体10;
3)将中间体10在氨的甲醇溶液中经乙酯基的氨解反应,制得中间体酰胺11;
4)中间体酰胺11在钠氢条件下完成C环分子内环合反应,制得目标产物12;
5)目标产物12经过NBS溴代反应或硝化反应,制得溴代取代物13a或硝基取代物13b;其中,中间体8的结构式如下:
中间体10的结构式如下:
中间体酰胺11的结构式如下:
目标产物12的结构式如下:
溴代取代物13a或硝基取代物13b的结构式如下:
其中,R为H,Br或NO2;n=1或2或3。
步骤1)中,邻氨基苯甲酰胺与草酸二乙酯的反应摩尔比为1:(6~8)。
步骤2)中,中间体8、碳酸钾及二溴代烷烃的反应摩尔比为1:(1.1~1.3):(1.1~1.3)。
步骤3)中,中间体10与氨的甲醇溶液的反应用量比为1g:(6~32)mL。
步骤4)中,中间体酰胺11与钠氢的反应摩尔比为1:(2~2.2)。
本发明还公开了上述2,3-内酰胺环稠合喹唑啉-4(3H)-酮衍生物在制备抗肿瘤药物中的应用。
所述的抗肿瘤药物为抗肝癌的药物。
所述的抗肝癌的药物为抑制人肝癌细胞SMMC-7721和Hep G2增殖的药物。
与现有技术相比,本发明具有以下有益的技术效果:
本发明公开的2,3-内酰胺环稠合喹唑啉-4(3H)-酮衍生物的制备方法,以邻氨基苯甲酰胺为起始原料,通过与草酸二乙酯稠合构建A、B环,得到中间体8,然后与二溴代烷烃在干燥的DMF中以碳酸钾为缚酸剂进行烷基化反应得到中间体10,中间体10在氨水的甲醇溶液中经乙酯基的氨解反应得到中间体酰胺,用于构建C环的分子内环合反应是在钠氢条件下完成,由此可得到目标产物,目标产物经过NBS溴代和硝化反应获得其他两个取代的目标产物。总之,本专利共合成了8个新化合物,包括4个目标化合物(12a,12b,13a和13b)和4个中间体(10a,10b,11a和11b)。该方法条件比较温和,易操作,后处理简单,非常实用,可推广运用于该类化合物的合成。因目前迫切需要结构多样化的喹唑啉杂环骨架结构,这种重要的骨架可以作为中间体用于进一步合成具有生物活性的化合物以研发更多有效的活性杂环化合物。除此之外,该法还可衍生用于合成其他类似的稠和杂环体系,如吴茱萸次碱的N-杂衍生物等。
本发明对合成得到的目标化合物进行了抗肝癌细胞生物活性测试,并研究了稠合杂环A环上的取代基对活性的影响,发现A环非取代的结构在抗肝癌细胞活性方面显示出独特的优势,比较硝基取代和溴代的衍生物活性较好。初步研究结果表明,化合物1其对易转染性肝癌细胞的抑制活性比对照组索拉非尼和舒尼替尼的活性好,表现出较好的先导化合物的势头,该研究结果具有很强的现实指导意义,可为进一步寻找新型结构的具有稠合喹唑啉环的抗肿瘤药物提供先导结构。
附图说明
图1为本发明2,3-内酰胺环稠合喹唑啉-4(3H)-酮目标化合物的合成路线图;
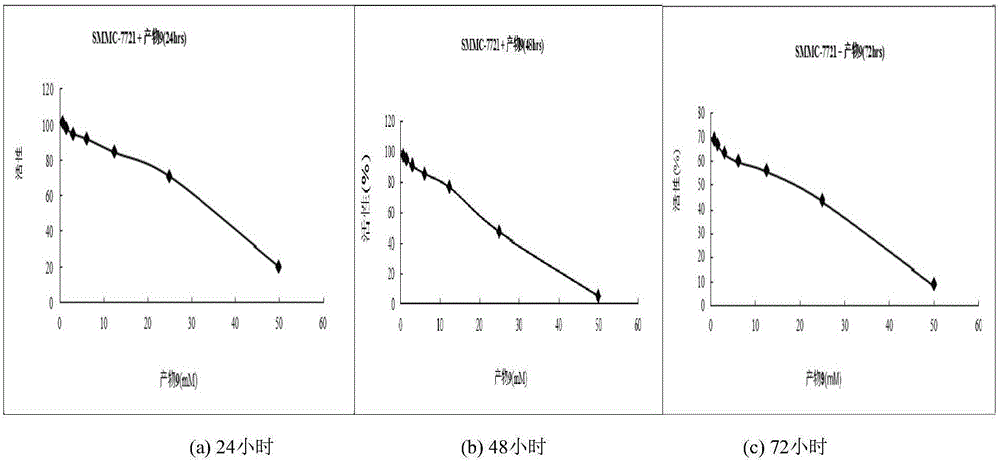
图2-1为化合物12a对SMMC-7721细胞的抑制活性结果图;
图2-2为索拉非尼对SMMC-7721细胞的抑制活性结果图;
图2-3为苏尼替尼对SMMC-7721细胞的抑制活性结果图;其中,(a)为24h;(b)为48h;(c)为72h;
图3为化合物12a、索拉非尼和苏尼替尼三种化合物的抑制活性对比结果;
图4为化合物10a的MS图谱;图5为化合物11a的MS图谱;
图6为化合物12a的MS图谱;图7为化合物10a的氢谱;
图8为化合物11a的氢谱;图9为化合物12a的氢谱;
图10为化合物10b的氢谱;图11为化合物11b的氢谱;
图12为化合物12b的氢谱。
具体实施方式
下面结合具体的实施例对本发明做进一步的详细说明,所述是对本发明的解释而不是限定。
一、本发明公开的2,3-内酰胺环稠合喹唑啉-4(3H)-酮衍生物的合成方法
本发明设计了一系列具有线性并环结构的B环稠合的喹唑啉-4-酮衍生物可作为拓扑异构酶抑制剂或酪氨酸激酶抑制来获得更好抗肿瘤活性,以商业易得的邻氨基苯甲酰胺和草酸二乙酯为起始原料,成功合成了这三环稠合的喹唑啉-4-酮衍生物,并对其进行结构衍生化,制备了系列目标化合物,该系列化合物保留了上市替尼类抗肿瘤新药喹唑啉环的基本活性骨架,同时简化了天然产物拓扑异构酶抑制剂(如喜树碱和骆驼宁碱等)的多环稠合结构,由此可以简化目标产物的合成路线和工艺,本专利公开的这种新型三环结构化合物的合成方法未见文献报道。目标化合物的合成路线如图1所示。
该目标产物的稠和杂环A、B、C环是依次通过4~5步化学反应构建起来的,具体合成方法是以邻氨基苯甲酰胺(6)为起始原料,通过与草酸二乙酯(7)稠合构建A、B环,得到中间体8,然后与二溴代烷烃(9a~9c)在干燥的DMF中以碳酸钾为缚酸剂进行烷基化反应得到中间体10,化合物10在氨水的甲醇溶液中经乙酯基的氨解反应得到中间体酰胺11,用于构建C环的分子内环合反应是在钠氢条件下完成,由此可得到目标产物12,化合物12a经过NBS溴代和硝化反应获得其他两个取代的目标产物13a和13b。总之,本专利共合成了8个新化合物,包括4个目标化合物(12a,12b,13a和13b)和4个中间体(10a,10b,11a和11b)。通过实验研究还发现,在稠合喹唑啉环上引入吸电子基会导致产率大大降低,越早引入吸电子基产率越低,研究路线中以在最后一步引入取代基产率最高。通过对肝癌细胞活性测定,可得产物3,4-二氢-9-三氮杂蒽-1,10-二酮(1a)比较阳性药索拉菲尼和舒尼替尼具有较好的癌细胞抑制活性。
具体实施例如下:
实施例1:4-氧代-3,4-二氢喹唑啉-2-羧酸乙酯(8)的合成
于250毫升的三颈瓶中加入邻氨基苯甲酰胺(6,6.8g,50mmol)和草酸二乙酯(7,50mL,367mmol)及磁子,加热搅拌,此时温度为140℃,由澄清的浅黄色变为浑浊的橘红色,一小时后于150℃下开始回流,五小时后变为黄白色,48小时后薄层层析(TLC)监控,产物无荧光,表示反应进行完全。冷却至室温,即析出固体。抽滤,用无水乙醇洗得到滤饼。将所得滤饼用无水乙醇重结晶,所得产物在真空条件下干燥得白色针状细小晶体为所需中间体8(4.928g,44.8%),m.p.185.8~187.4℃。
实施例2:4-氧代-3,4-二氢喹唑啉-2-羧酸乙酯(8)的合成
将草酸二乙酯(7,50mL,0.366mol)慢慢小心地分批量加入融化的2-氨基苯甲酰胺(6,6.8g,0.05mol)之中。将反应混合物搅拌下加热回流48小时,之后用薄层层析(TLC)监控(展开剂:DCM–Et2O,v:v=1:1)反应进程。确定反应完全后将反应液冷却至室温,将过量的草酸二乙酯减压下蒸馏除去。所得固体用冷的乙醇(100mL)洗涤,抽滤后得到滤饼再悬浮于冷的乙醚(50mL)中,搅拌0.5小时,过滤得到所需中间体(8,8.6g,78.9%)呈细小白色针状晶体,m.p.190–191℃。
实施例3:4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(8)的合成
搭好回流装置,于250mL的三颈瓶中加入邻氨基苯甲酰胺(6,6.8g,50mmol)和草酸二乙酯(7,50mL,367mmol)及磁子,加热搅拌,此时温度为140℃,由澄清的浅黄色变为浑浊的橘红色,一小时后于150℃下开始回流,5小时后变为黄白色,反应48小时后TLC监控,产物无荧光或荧光点,表示反应进行完全。冷却至室温,即析出固体。抽滤得滤饼,用无水乙醇洗。用无水乙醇重结晶,真空干燥得所需中间产物(8,5.84g,53.1%),m.p.186–187℃。
实施例4:3-(2-溴乙烷基)-4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(10a)的合成
在500mL反应瓶中加入4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(8,3.068g,14mmol)及DMF(250mL),搅拌至澄清溶解后加入K2CO3(7.352g),量取1,2-二溴乙烷(9a,8.001g,44mmol),滴加进该反应体系中。反应过夜,TLC监测反应,21小时后TLC监测反应进行完全。抽滤,滤掉K2CO3,加入水(1000mL),用乙酸乙酯(1000mL)萃取三次,直至水层用TLC监测在紫外下不显色,萃取完毕。用无水Na2SO4干燥有机层。旋蒸至黄色油状且无乙酸乙酯旋出。加少量乙醇和适量的水,析出白色固体,过滤,干燥的白色粉末即所需中间产物(10a,4.213g,91.8%),m.p.69.4~73.0℃。1H NMR(400MHz,CDCl3)δ:8.32(d,J=7.9Hz,1H),7.81(d,J=3.5Hz,2H),7.59(dd,J=7.7,4.3Hz,1H),4.64–4.45(m,4H),3.86(t,J=6.5Hz,2H),1.48(t,J=7.1Hz,3H).MS m/z calculated for C13H13BrN2O3[M+H]+325.16,found326.1。
实施例5:3-(2-溴乙烷基)-4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(10a)的合成
将4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(8,2.18g,10.0mmol)和碳酸钾(1.52g,11.0mmol)在干燥DMF(50mL)中,在室温和充分搅拌条件下,在30分钟内滴加1,2-二溴乙烷(9a,2.06g,11mmol),反应混合物在室温条件搅拌反应4个小时。反应进程用TLC监控,当反应完全后,过滤除去碳酸钾,并向滤液中加入水(200mL)后将产物用乙酸乙酯(3×100mL)萃取。合并有机相用无水硫酸钠干燥,旋转蒸发得到粗产物为淡黄色油状物,用柱层析色谱分离纯化,展开剂为石油醚:乙酸乙酯(v/v=5:1),得到无色油状物,加入少量的乙醇和适量的水,超声条件下析出大量白色固体为所需中间体10a(2.5g,76.9%).m.p.73-74℃;1H NMR(400MHz,CDCl3)δ:8.32(d,J=7.9Hz,1H),7.81(d,J=3.5Hz,2H),7.59(dd,J=7.7,4.3Hz,1H),4.64–4.45(m,4H),3.86(t,J=6.5Hz,2H),1.48(t,J=7.1Hz,3H).MS m/z calculated for C13H13BrN2O3[M+H]+325.16,found 326.1。
实施例6:3-溴乙烷基-4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(10a)的合成
在500mL单口烧瓶中,加入4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(8,1.23g,5.6mmol)及DMF(35mL),搅拌至澄清溶解后加入K2CO3(1.01g,7.3mmol),量取二溴乙烷(1.4g,7.3mmol),滴加进体系中。反应过夜,TLC监测反应,21小时后TLC监测反应进行完全。抽滤,滤掉K2CO3,加入水(150mL),用乙酸乙酯(150mL)萃取三次,水层点在点板上无点,萃取完毕。用无水Na2SO4干燥有机层。旋蒸至黄色油状且无乙酸乙酯旋出。加少量乙醇和适当水,析出白色固体,过滤,干燥的白色粉末即为所需中间产物(10a,1.90g,90.7%),m.p.73-74℃;1H NMR(400MHz,CDCl3)δ:8.32(d,J=7.9Hz,1H),7.81(d,J=3.5Hz,2H),7.59(dd,J=7.7,4.3Hz,1H),4.64–4.45(m,4H),3.86(t,J=6.5Hz,2H),1.48(t,J=7.1Hz,3H).MS m/z calculated for C13H13BrN2O3[M+H]+325.16,found 326.1。
实施例7;3-(3-溴丙烷基)-4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(10b)的合成
将4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(8,2.18g,10.0mmol)和碳酸钾(1.52g,11.0mmol)加入干燥DMF(50mL)中,在室温下充分搅拌,在30分钟内滴加1,3-二溴丙烷(9b,2.21g,11.0mmol),反应混合物在室温条件搅拌反应6个小时。反应进程用TLC监控,当反应完全后,过滤除去碳酸钾,并向滤液中加入水(200mL)后,将产物用乙酸乙酯(3×100mL)萃取。合并有机相用无水硫酸钠干燥,减压旋转蒸发得到粗产物为淡黄色油状物,用柱层析色谱分离纯化,展开剂为石油醚:乙酸乙酯(v/v=5:1),得到无色油状物,加入少量的乙醇和适量的水,超声条件下析出大量黄色固体为所需中间体10b(0.97g,28.6%),m.p.62.8-64.0℃;1H NMR(400MHz,CDCl3)δ:8.32(d,J=7.9Hz,1H),7.81(d,J=3.5Hz,2H),7.59(d t,J=11.6,3.9Hz,1H),4.55(m,4H),3.86(t,J=6.5Hz,2H),1.48(t,J=7.1Hz,3H)。
实施例8:3-溴甲基-4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(10c)的合成
将4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(8,2.18g,10.0mmol)和碳酸钾(1.52g,11.0mmol)加入干燥DMF(50mL)中,在室温下充分搅拌,在30分钟内滴加二溴甲烷(9c,1.91g,11.0mmol),反应混合物在室温条件搅拌反应8个小时。反应进程用TLC监控,当反应完全后,过滤除去碳酸钾,并向滤液中加入水(200mL)后,将产物用乙酸乙酯(3×100mL)萃取。合并有机相用无水硫酸钠干燥,减压旋转蒸发得到粗产物为淡黄色油状物,用柱层析色谱分离纯化,展开剂为石油醚:乙酸乙酯(v/v=5:1),得到无色油状物,加入少量的乙醇和适量的水,超声条件下析出大量黄色固体为所需中间体10c(0.96g,28.1%)。HR MS m/z calculated for C12H11BrN2O3[M+H]+.309.9953,found 309.9956。
实施例9:3-(2-溴乙基)-4-氧代-3,4-二氢-喹唑啉-2-甲酰胺(11a)的合成
取3-溴乙烷基-4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(10a,4.21g,13.0mmol),加入氨的甲醇溶液(7N,50mL),溶液由澄清变为乳白色浑浊。于室温下搅拌过夜,20小时后TLC监测反应进行完全。加入冰水(250mL)并搅拌10分钟。加浓盐酸调节pH值至5到6。抽滤,干燥。得白色粉末产物11a(3.3g,86.7%),m.p.74.9~76.8℃.1H NMR(400MHz,CDCl3)δ:8.32(d,J=8.0Hz,1H),7.81(t,J=7.6Hz,1H),7.72(d,J=8.1Hz,1H),7.59(t,J=7.5Hz,1H),7.52(s,1H),5.84(s,1H),4.97(t,J=6.5Hz,2H),3.90(t,J=6.5Hz,2H).HRMS m/z calculated for C11H10BrN3O2[M+H]+.294.9956 296.12,found 294.9952。
实施例10:3-(2-溴乙烷基)-4-氧代-3,4-二氢-喹唑啉-2-甲酰胺(11a)的合成
将中间体10a(1.30g,4.0mmol)至于一个50mL的圆底烧瓶中,在充分搅拌条件下向其中分批加入氨的甲醇溶液(7N,8.0mL),反应混合物在室温条件下搅拌过夜(大约16个小时)。反应进程用TLC监控分析。待反应完全后,将反应所得悬浮混合物倾入冰水(40mL)中搅拌10分钟。用浓盐酸调节pH值=5~6。得到大量白色沉淀,抽滤用水洗涤滤饼,真空干燥得到所需产物为11a(1.04g,88.14%).m.p.171-173℃1H NMR(400MHz,CDCl3)δ:8.32(d,J=8.0Hz,1H),7.81(t,J=7.6Hz,1H),7.72(d,J=8.1Hz,1H),7.59(t,J=7.5Hz,1H),7.52(s,1H),5.84(s,1H),4.97(t,J=6.5Hz,2H),3.90(t,J=6.5Hz,2H).MS(EI)m/z calculated for C11H10BrN3O2[M+H]+.296.12,found 216.1(MS-Br)。
实施例11:3-溴乙烷基-4-氧代-3,4-二氢-喹唑啉-2-羧酸乙酯(11a)的合成
取产物10a(1.90g,5.8mmol),加入氨的甲醇溶液(7N,60mL),溶液由澄清变为乳白色浑浊。于室温下搅拌过夜,20小时后TLC监测反应进行完全。加入冰水(3000mL)并搅拌10分钟。加浓盐酸调节pH值至5到6。抽滤,干燥。得白色粉末产物11a(1.67g,86.7%),波谱分析鉴定结果同上。
实施例12:3-(3-溴丙烷基)-4-氧代-3,4-二氢-喹唑啉-2-甲酰胺(11b)的合成
将中间体10b(1.35g,4.0mmol)至于一个50mL的圆底烧瓶中,边搅拌边向其中分批加入氨的甲醇溶液(7N,8.0mL),反应混合物在室温条件下搅拌过夜(大约16个小时)。反应进程用TLC监控分析。待反应完全后,将反应所得悬浮混合物倾入冰水(40mL)中搅拌10分钟。用浓盐酸调节pH值=5~6。得到大量白色沉淀,抽滤用水洗涤滤饼,真空干燥得到所需产物为11b(1.0g,75.4%).m.p.146.0-147.0℃.1H NMR(400MHz,CDCl3)δ:8.32(d,J=8.0Hz,1H),7.81(t,J=7.6Hz,1H),7.72(d,J=8.1Hz,1H),7.59(t,J=7.5Hz,1H),7.52(s,1H),5.84(s,1H),4.97(t,J=6.5Hz,2H),3.90(t,J=6.5Hz,2H)。
实施例13:3,4-二氢-9-三氮杂蒽-1,10-二酮(12a)的合成
取产物11a(3.3g,11.0mmol),加入THF(100mL)使产物11a完全溶解。量取NaH(0.88g,22mmol),在冰浴条件下(-5~0℃)缓慢加入NaH。溶液由澄清变为乳白色。将体系移至室温环境下搅拌反应1小时。TLC监测待反应进行完全。减压浓缩后,加入(150mL)冰水,过滤,用冰水和石油醚洗涤,直到洗白。称量得产物12a(2.28g,95.3%),m.p.>300℃。1H NMR(400MHz,DMSO-d6)δ:8.91(s,1H),8.18(d,J=7.7Hz,1H),7.89(t,J=7.2Hz,1H),7.82(d,J=8.0Hz,1H),7.63(t,J=7.4Hz,1H),4.22(m,2H),3.55(t,2H).MS m/z calculated for C11H9N3O2[M+H]+215.21,found 215.1。
实施例14:3,4-二氢-9-三氮杂蒽-1,10-二酮(12a)的合成
将中间体11a(0.89g,3.0mmol)溶解于THF(25mL)之中,在-5-0℃条件下向其中慢慢加入NaH(0.24g,6.0mmol,60%)。之后将反应混合物在室温下搅拌1小时。反应进程用TLC监控,待反应完全后将混合物减压浓缩后倒入冰水(50mL)之中,过滤得到粗产物,用冷水、石油醚依次洗涤滤饼得到所需终产物12a(0.56g,86.8%),m.p.>300℃1H NMR(400MHz,DMSO-d6)δ:8.91(s,1H),8.18(d,J=7.7Hz,1H),7.89(t,J=7.2Hz,1H),7.82(d,J=8.0Hz,1H),7.63(t,J=7.4Hz,1H),4.22(m,2H),3.55(t,2H).MS m/z calculated for C11H9N3O2[M+H]+215.21,found 215.1。
实施例15:3,4-二氢-9-三氮杂蒽-1,10-二酮(12a)的合成
取产物11a(1.67g,5.70mmol),加入THF(60mL)使产物11a完全溶解。量取NaH(0.29g,12mmol),在冰浴条件下(-5~0℃)缓慢加入NaH。溶液由澄清变为乳白色。将体系移至室温环境下搅拌反应1小时。TLC监测反应进行完全。旋蒸。加入冰水(120mL),过滤,用冰水和石油醚洗涤,直到洗白得产物12a(1.14g,90.2%),1H NMR(400MHz,DMSO-d6)δ:8.91(s,1H),8.18(d,J=7.7Hz,1H),7.89(t,J=7.2Hz,1H),7.82(d,J=8.0Hz,1H),7.63(t,J=7.4Hz,1H),4.22(m,2H),3.55(t,2H).MS m/z calculated for C11H9N3O2[M+H]+215.21,found 215.1。
实施例16:2,3,4,5-四氢-[1,4]二氮杂[2,1-b]喹唑啉-1,7-二酮(12b)的合成
将化合物11b(0.47g,1.50mmol)溶解于THF(10.0mL)之中,在-5-0℃条件下向其中慢慢加入NaH(0.12g,3.0mmol,60%)。反应混合物在室温条件下搅拌3小时。反应进程用TLC监控,待反应完全后将混合物减压浓缩后所得残余物倒入冰水(50mL)中,过滤得到粗产品,依次用冷水、石油醚依次洗涤滤饼,得到所需终产物12b(0.12g,35.0%yield).m.p.>300℃.1H NMR(400MHz,DMSO-d6)δ:8.91(s,1H),8.18(d,J=7.7Hz,1H),7.89(t,J=7.2Hz,1H),7.82(d,J=8.0Hz,1H),7.63(t,J=7.4Hz,1H),4.22(m,2H),3.55(t,2H)。
实施例17:6-硝基-3,4-二氢-9-三氮杂蒽-1,10-二酮(13b)的合成
硝基取代稠合喹唑啉环的最佳合成路线是以邻氨基苯甲酰胺为起始原料通过与草酸二乙酯稠合,二溴乙烷的烷基化反应,氨解,环合合成稠合喹唑啉环,最后在稠合喹唑啉环上添加硝基取代基。具体合成方法如下:取产物12a(2.286g,10.6mmol),加入浓硫酸(40.0mL),量取浓硝酸(8.0mL)置于恒压漏斗中。缓慢滴加浓硝酸到反应体系中。4小时后TLC监测,反应进行完全。加入碎冰(400g),立即洗出浅黄色固体。抽滤得粗产品(1.06g,m.p.231.7~大于300℃)。进而采用制备薄层层析对粗品进行纯化,得最终产物为硝基取代稠合喹唑啉环产物13b(0.927g,m.p.162.2~165.7℃,25.6%)。
实施例18:6-溴-3,4-二氢-9-三氮杂蒽-1,10-二酮(13a)的合成
溴取代的喹唑啉三环衍生物13a的最佳合成路线也是以邻氨基苯甲酰胺为起始原料通过与草酸二乙酯稠合,与二溴乙烷反应进行溴烷基化,再经氨解后发生分子内环合的形成稠环喹唑啉产物,最后在三环骨架上引入溴原子得到目标产物。具体合成方法如下:量取NBS(1.03g,5.5mmol)与500mL圆底烧瓶中,于冰浴中0℃条件下加入干燥的DMF(100mL),取化合物12a(1.14g,5.30mmol)置入该混合物中。搅拌1小时。TLC监测反应进行完全。旋蒸,过夜。抽滤后加入饱和Na2CO3,用乙酸乙酯萃取。有机层加入无水硫酸钠,干燥过夜。抽滤旋干得粗品。经过柱层析纯化得固体产物为溴代喹唑啉三环衍生物13a(1.061g,44.7%),m.p.201.7~207.4℃。
本发明中所涉及到的新化合物的编号和结构如表1所示。
表1.本专利中所涉及到的新化合物的编号和化学结构
本发明制得的新化合物的波谱鉴定结果如附图4~图12所示。
二、目标化合物的肿瘤细胞抑制活性
药物抗肿瘤的毒理作用检测方法的具体方法步骤如下:
1.肝癌细胞系SMMC-7721和Hep G2以含10%胎牛血清的DMEM培养基(Corning cat no.10017236R),37℃、5%CO2的培养箱培养至对数生长期。以0.02%EDTA-0.25%胰酶37℃消化两分钟,1000rpm离心5分钟收集细胞。
2.制备1*10^5/mL细胞悬液,按100μL/孔将细胞接种于96孔细胞培养板,37℃、5%CO2过夜培养细胞生长至对数期。
3.将所测试的药物(12a,12b,13a,13b等)以DMSO配置为100mM贮存液,分别按0-50mM系列浓度梯度进行加药处理,均设三个复孔,总体积200μL/孔,继续37℃、5%CO2下培养细胞24h、48h、72h。
4.加入20μL/孔MTT(5mg/mL),继续培养3-4h,活细胞内将形成蓝紫色不溶物甲躜。
5.移除培养液,加150μL/孔DMSO,室温下振荡15分钟,使甲躜充分溶解。
6.在490nm波长酶标仪测定相对吸光度,根据下列公式得到细胞存活率曲线图及药物半数致死剂量(IC50):细胞生长存活率=实验组的OD490nm/对照组的OD490nm*100%
采用上述方法测试化合物的活性,分别在24小时、48小时、72小时的时候,测定并记录产物对SMMC-7721细胞抑制活性。以活性最好的化合物12a为例,将所得活性测试结果总结在表2之中。
表2.化合物12a分别在24/48/72小时对SMMC-7721细胞的抑制活性测定数据
化合物12a在24小时,48小时,72小时产物9对SMMC-7721细胞的IC50值比较如表3所示:
表3.半数抑制浓度对比
另外,采用对照品索拉非尼及苏尼替尼与药物活性测试评价同时平行展开,分别在24小时、48小时、72小时,测试其对SMMC-7721细胞的抑制活性,数据结果如表4所示:
表4.索拉非尼和苏尼替尼在24小时对SMMC-7721的抑制活性表
对化合物12a,苏尼替尼、索拉非尼三种化合物分别在24小时/48小时/7小时对SMMC-7721细胞抑制活性的IC50值进行对比,结果如表5所示:
表5.三种化合物在24小时/48小时/72小时对SMMC-7721细胞抑制活性的IC50值对比
对以上活性数据进行总结绘图。化合物12a对SMMC-7721细胞的抑制活性(24小时、48小时、72小时),结果参见图2-1;索拉非尼对SMMC-7721的细胞抑制活性(24小时、48小时、72小时),结果参见图2-2;苏尼替尼的细胞抑制活性(24小时,48小时,72小时),结果参见图2-3。将苏尼替尼、索拉非尼和化合物12a的活性对比,结果参见图3,可得出产物12a显示出有效的抗肿瘤活性,具有良好的研究前景。化合物12a能有效抑制人肝癌细胞(SMMC-7721和Hep G2)的增殖,其机制与Bax/Bcl-2比值的上调和Cyclin D1转录水平的下调有关,说明化合物GWJ具有抗肝癌的作用。尤其是化合物12a对具有较高转染性的抗耐药性肝癌细胞株SMMC-7721显示出比阳性药索拉非尼和舒尼替尼较强的抑制活性,说明该化合物具有较好研发前景。
本发明以天然产物骆驼宁碱A中所含稠合喹唑啉环为模板,设计合成了一系列新型结构的简化的三环稠合喹唑啉衍生物,并对其进行了抗肝癌细胞的活性测试实验,化合物12a比阳性药舒尼替尼和索拉菲尼表现出较好的抑制活性和较低的IC50值,该结果为骆驼宁碱和喜树碱的结构改造提供了一定研究方向,并可为进一步发现抗肿瘤药物提供新型的先导结构。本发明所用于合成该类稠合三环喹唑啉衍生物的方法经过多次试验验证,涉及了连续的4~5步传统的化学反应(分子间环合、烷基化、氨解、分子内环合与取代反应),并从多方面证实这种反应序列的安排最有利于终产物产率的提高,整个合成路线用于该类化合物的合成首次被公开报道,可为该类衍生物的大量合成和结构改造提供参考方法。
- 还没有人留言评论。精彩留言会获得点赞!