艾沙康唑一水合物晶型及其制备方法与流程
本发明属于药学领域,具体的涉及一种艾沙康唑一水合物晶型及其制备方法。
背景技术:
硫酸艾沙康唑鎓(Isavuconazonium sulfate)由安斯泰来(Astellas,日本)和巴塞利亚(Basilea,瑞士)联合开发,FDA已授予其合格传染病产品(QIDP)资格认定的氮唑类抗真菌药物。2014年7月8日安斯泰来向FDA提交了上市申请,2015年3月获批。硫酸艾沙康唑鎓是一种用于治疗侵袭性曲霉菌感染和侵袭性毛霉菌感染的前体药物。
在血液中,前体药物在酯酶(主要为丁酰胆碱酯酶)的作用下迅速水解为活性物质艾沙康唑。艾沙康唑的作用机制与其他氮唑类抗真菌药物类似,其作用机理主要是减少麦角甾醇的合成而发挥作用。麦角甾醇是真菌细胞合成过程中必不可少的物质,其参与细胞上一些重要蛋白质的合成,是真菌细胞中必须的物质。当艾沙康唑作用于真菌细胞后,与真菌内羊毛甾醇14α-去甲基化酶(P45014DM)相竞争作用,降低其活性,使细胞内羊毛甾醇积累而麦角甾醇缺少,导致细胞膜无法合成,从而发挥艾沙康唑的药效。
硫酸艾沙康唑鎓的制备过程需要用到的一个必须的关键中间体,也是其体内水解后的活性成分—艾沙康唑,艾沙康唑的化学名称为:4-[2-[(1R,2R)-2-(2,5-二氟苯基)-2-羟基-1-甲基-3-(1H-1,2,4-三唑-1-基)丙基]-4-噻唑基]苯甲腈,其结构式6如下所示:
根据当前国内外专利和文献资料报道,艾沙康唑本身为无定形白色粉末(专利国际申请:WO2016055918A1中第3页提到“a solid amorphous form”)、或者为无色浆状物或者粘稠状物(专利国际申请:WO1999045008A1中第28页提到“colorless heavy syrup”)。专利国际申请:WO2016055918A1中还提到了一种艾沙康唑氢溴酸盐的晶型或者通过游离艾沙康唑氢溴酸盐的方法制备出三种艾沙康唑的晶型,但是这些晶型的制备方法均需要经过艾沙康唑氢溴酸盐来制备,制备工艺复杂且其晶型未涉及水合物。由于无定形粉末、浆状物或者非水合物在制备过程中没有晶体析出的过程,在杂质和溶剂残留去除方面都存在较大困难。在实际制备过程中,无定形粉末在析出固体时容易发粘,存在严重的挂壁、贴壁、团聚等现象,过程工艺复杂、耗时较长,这都是该无定形粉末制备工艺在工业化生产中无法突破的技术壁垒。本领域渴望一种新的艾沙康唑晶型,其与以往的艾沙康唑无定形粉末或晶型相比,具有结晶工艺简单、可操作性强、稳定性好等优点,更有利于工业化生产。
技术实现要素:
本发明人采用(2S,3R)-3-(2,5-二氟)-3-羟基-2-甲基-4-(1H-1,2,4-三唑-1-基)丁腈、二硫代磷酸二乙酯、2-溴-4氰基-苯乙酮为原料,经过加成反应、关五元杂环的步骤以及独特的后处理和结晶工艺制备出艾沙康唑一水合物白色结晶性固体粉末。该工艺制备出的艾沙康唑一水合物晶体粉末与以往的艾沙康唑无定形粉末相比,具有结晶工艺简单、可操作性强、稳定性好等优点,更有利于工业化生产。
本发明的目的是提供一种艾沙康唑一水合物晶型。
本发明的第二个目的是提供艾沙康唑一水合物晶型的制备方法。
在本发明的实施方案中,本发明提供了一种新的4-[2-[(1R,2R)-2-(2,5-二氟苯基)-2-羟基-1-甲基-3-(1H-1,2,4-三唑-1-基)丙基]-4-噻唑基]苯甲腈一水合物晶型,也就是艾沙康唑的水合物晶型。
在本发明的优选实施方案中,本发明提供的艾沙康唑一水合物是结晶形式的,更优选地,其XRPD图谱中2θ值在7.3±0.2,13.5±0.2,;14.0±0.2,14.5±0.2,15.7±0.2,16.1±0.2,17.3±0.2,19.5±0.2,19.9±0.2,20.5±0.2,21.3±0.2,21.9±0.2,22.2±0.2,23.6±0.2,24.2±0.2,24.7±0.2,26.0±0.2,26.6±0.2,27.2±0.2,27.8±0.2,28.3±0.2,29.3±0.2,29.9±0.2,31.9±0.2,32.4±0.2,33.8±0.2,34.4±0.2,36.7±0.2,38.0±0.2处有衍射峰。
在本发明的更优选实施方案中,本发明提供的艾沙康唑一水合物晶型,其XRPD图谱基本如图1所示。
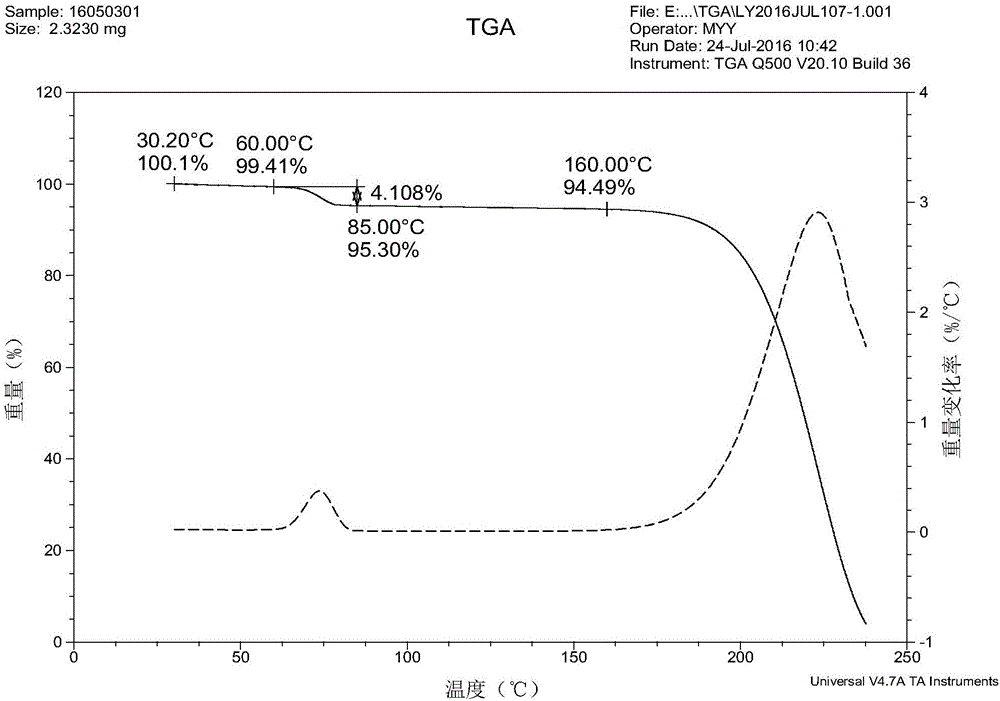
在本发明的更优选实施方案中,本发明提供的艾沙康唑一水合物晶型,其TGA图谱在60℃-85℃之间约有4.1%w/w的台阶式失重(为结晶水解离失重平台),在约160℃之后开始逐渐分解,为一水合物晶型;优选地,其TGA图谱基本如图2所示。
在本发明的更优选实施方案中,本发明提供的艾沙康唑一水合物晶型,其DSC图谱在约74.73℃有吸热转变,特别优选地,其DSC图谱基本如图3所示。
第二方面,本发明提供了上述艾沙康唑的一水合物晶型的制备方法,所述方法包括以下步骤:
(1)式1化合物与产生H2S气体的试剂反应,得到式3化合物;
(2)式3化合物与2-溴-4氰基-苯乙酮发生关五元杂环反应,再经过调节pH值、有机溶剂打浆以及混合溶剂结晶后,得到式5化合物晶型,即艾沙康唑一水合物晶型。
在本发明的实施方案中,本发明提供的艾沙康唑的一水合物晶型的制备方法,其中,步骤(1)为式1化合物与产生H2S气体的试剂反应,使式1化合物结构中的氰基与H2S发生加成反应,所述产生H2S气体的试剂可选自二硫代磷酸二乙酯或者Na2S,优选的为二硫代磷酸二乙酯。并且,在与水互溶的有机溶剂中,在少量水存在的条件下,发生加成反应,得到式3化合物,这里,所述与水互溶的有机溶剂选自异丙醇、乙醇、甲醇、四氢呋喃、乙腈等,优选地为异丙醇。
在本发明的优选实施方案中,本发明提供的艾沙康唑的一水合物晶型的制备方法,其中,步骤(1)中所述的产生H2S气体的试剂采用的是二硫代磷酸二乙酯,以及采用异丙醇做溶剂,式1化合物与二硫代磷酸二乙酯的摩尔比为1:1-1:6,式1化合物与水的摩尔比为1:1-1:50,加成反应的温度为20℃-100℃,反应时间为1-48h;进一步优选地,式1化合物与二硫代磷酸二乙酯的摩尔比为1:4,式1化合物与水的摩尔比为1:10,加成反应的温度为70℃-90℃,反应时间为6-15h。
在本发明的实施方案中,本发明提供的艾沙康唑的一水合物晶型的制备方法,其中,步骤(1)在反应完毕后采用本领域常规的方法进行后处理纯化,例如,采用无机碱调节pH至中性至弱碱性,优选地,在搅拌下慢慢滴加10wt%NaOH溶液调节pH值到7-8。调节pH后采用与水不互溶的溶剂萃取分层,合并有机相,优选地,采用二氯甲烷萃取分层,合并二氯甲烷相后减压浓缩。减压浓缩后采用加混合溶剂搅拌析出固体的方法结晶并纯化,优选地,所述混合溶剂为二氯甲烷和正庚烷的混合溶剂,二氯甲烷和正庚烷体积比为1:3。
在本发明的实施方案中,本发明提供的艾沙康唑的一水合物晶型制备方法,其中,步骤(2)为:式3化合物与2-溴-4氰基-苯乙酮发生关五元杂环反应,经过调节pH值、有机溶剂打浆以及混合溶剂结晶后,得到式5化合物晶型,即艾沙康唑一水合物晶型。这里,所述关五元杂环反应在溶剂存在条件下进行,所用溶剂为乙醇、甲醇、乙腈、四氢呋喃、异丙醇、二氯甲烷、或乙酸乙酯等有机溶剂。式3化合物与2-溴-4氰基-苯乙酮的摩尔比为1:0.5-1:5,反应温度为10℃-120℃,所述关五元杂环反应的时间为1-24h。优选地,所述关五元杂环反应所用的溶剂为乙醇,而且任选地所述乙醇为含水的;式3化合物与2-溴-4氰基-苯乙酮的摩尔比为1:1-1:1.2,所述关五元杂环反应的温度为40℃-80℃,反应时间为1-5h。
在本发明的优选实施方案中,本发明提供的艾沙康唑的一水合物晶型制备方法,其中步骤(2)中所述关五元杂环反应所用的溶剂为95体积%的乙醇水溶液;式3化合物与2-溴-4氰基-苯乙酮的摩尔比为1:1,关五元杂环反应的温度为80℃,反应时间为1-2h。
在本发明的实施方案中,本发明提供的艾沙康唑的一水合物晶型制备方法,其中步骤(2)反应完毕后,调节pH值步骤中,采用选自NaHCO3、Na2CO3、NaOH、Et3N等的无机碱或者有机碱,将pH调节至3-6范围内;优选地为采用NaHCO3调节pH值至4-5。
在本发明的实施方案中,本发明提供的艾沙康唑的一水合物晶型制备方法,其中步骤(2)反应完毕后,有机溶剂打浆工艺中,采用醚类、或酯类等有机溶剂打浆,打浆时间为1-10h。优选地为采用异丙醚打浆,打浆时间为2-6h。
在本发明的实施方案中,本发明提供的艾沙康唑的一水合物晶型制备方法,其中步骤(2)反应完毕后,结晶采用EtOH、MeCN、MeOH、THF等有机良溶剂与水等不良溶剂组成的混合溶剂进行搅拌结晶,结晶温度控制在-20-40℃;优选地,先采用EtOH和水的混合溶剂,后采用MeCN和水的混合溶剂进行结晶,结晶温度控制在15-25℃范围内。EtOH和水的混合溶剂中EtOH和水的体积比为1:0.5-1:3,优选地为1:1-1:2。MeCN和水的混合溶剂中MeCN和水的体积比为1:0.5-1:5,优选地为1:1-1:3。
在本发明的实施方案中,本发明提供的艾沙康唑的一水合物晶型制备方法,其中步骤(2)在反应完毕后的结晶步骤中,采用先乙醇或乙腈溶解后滴加水的方式,优选地,乙醇和水结晶的工艺为先使用乙醇将式5化合物溶解,搅拌下缓慢滴加水;乙腈和水结晶的工艺为先使用乙腈将式5化合物溶解,搅拌下缓慢滴加水。
在以往的专利和文献报道中,艾沙康唑多为无定形粉末或浆状物、或者为无色浆状物或者粘稠状物。在实际的工艺开发过程中,我们发现,无定形粉末、浆状物或者非水合物在制备过程中没有晶体逐渐析出的过程,其真实过程为先形成油状物,后油状物贴壁粘连,搅拌过程中形成贴壁的粘稠胶状物,再逐渐硬化继而固化,因此这种提纯过程在杂质和溶剂残留去除方面都存在较大困难。工业化生产中如果存在严重的挂壁、贴壁、团聚等现象,会增加过程工艺的复杂性、耗时较长,这都是无定形粉末制备工艺在工业化生产中无法实现的技术壁垒。另有专利国际申请:WO2016055918A1中还提到了一种艾沙康唑氢溴酸盐的晶型或者通过游离艾沙康唑氢溴酸盐的方法制备出三种艾沙康唑的晶型,但是这些晶型制备方法均需要经过艾沙康唑氢溴酸盐来制备,制备需要通过酸碱调节操作,工艺复杂。
本发明方法制备的一种新的艾沙康唑晶型,与以往的艾沙康唑无定形粉末或晶型相比,具有结晶工艺简单、可操作性强等优点,更有利于工业化生产。
附图说明
图1表示的是本发明实施例2得到的艾沙康唑的一水合物晶型的示例性XRPD图谱。
图2表示的是本发明实施例2得到的艾沙康唑的一水合物晶型的示例性TGA图谱。
图3表示的是本发明实施例2得到的艾沙康唑的一水合物晶型的示例性DSC图谱。
具体实施方式
下面通过实施例来进一步描述本发明的技术方案,这些实施例为示例性的,不构成对本发明保护范围的限定。本领域的技术人员,在本发明的教导下,根据现有技术而对其中技术特征进行等同替换仍属于本发明的保护范围内。
以下实施例中的式1化合物、式3化合物购自湖南欧亚,式2化合物购自上海希仕化学,其他原料和试剂,如无特别说明,均为普通市售产品。
本发明实施例中的英文缩写的含义:
DCM:二氯甲烷,昆山金城化工助剂厂,工业级。
EtOH:乙醇,国产工业级。
MeCN:乙腈,国产工业级。
EA:乙酸乙酯,国产工业级。
MeOH:甲醇,国产工业级。
EtOH:乙醇,国产工业级。
Heptane:正庚烷,国产工业级。
XRPD检测仪器以及检测条件:
Bruker D8Advance X-ray Diffractometer
1、Anode:Cu,2、Wavelegth1:1.5406A°,Wavelegth1:1.54443A°,3、Generator:40kV/40mA,4、Step:0.02°/3s,5、Start Angle:3°,End Angle:40°,6、Slit:1.0/1.0/Ni/0.2.
TGA检测仪器以及检测条件:
TA Q500型热重分析仪(TGA)
Ramp 10.0℃/min to 240.0℃,N2流为40mL/min。
DSC检测仪器以及检测条件:
TA Q200型差示扫描量热仪(DSC)
1、Equilibrate at 25.0℃,2、Ramp 10.0℃/min to 200.0℃3、N2流为40mL/min 4、铝盘,加盖
实施例1
式3化合物的制备
向5L三口圆底反应烧瓶中依次加入式2化合物,即二硫代磷酸二乙酯(804g)、式1化合物(300g)、异丙醇(1.5L)和纯化水(194ml),搅拌下升温至回流反应(油温85℃),内温78℃,保持回流,在升温过程中逐渐有大量H2S气体放出,直至温度稳定在内温78℃,无大量气体放出,保持温度搅拌。15h后,取样监测,TLC显示SM1点消失,新点生成。
降温至20℃以下,加入DCM(1.5L),纯化水(1.5L)。搅拌下慢慢滴加10wt%NaOH溶液(约2.3L)调节pH值到7-8,加入过程中体系变黄、变灰。分液萃取,收集下层DCM相。水相再用DCM(600ml)萃取两次。合并有机相,用饱和的NaCl溶液洗涤,用无水硫酸钠干燥。减压浓缩得到白色至淡黄色固体,加入DCM(300ml)和正庚烷(900ml),搅拌析晶17h。抽滤后,滤饼用DCM:正庚烷=1:6的溶液(300ml)洗涤,抽干后,真空旋干。收料得到318g白色固体,即式3化合物。收率94.5%。HPLC纯度99.68%。
实施例2
式5化合物的制备
向5L三口圆底反应烧瓶中依次加入实施例1制备的式3化合物(300g)、式4化合物即2-溴-4氰基-苯乙酮(215g)以及95%乙醇(1.5L),搅拌下升温至油温85℃,内温76℃,升温过程中体系颜色变黄绿色,保温搅拌2h。取样监测,TLC显示M1点消失,新点生成。
降温至20℃以下,搅拌下,将饱和的NaHCO3溶液缓慢的加入反应液中,调节pH值至4-5,有白色固体析出,继续搅拌至晶体完全析出后,再将水滴加至体系中,使总水量达到1.5L。搅拌2h后过滤,固体用EtOH:水=1:1的混合溶剂洗涤。将所得固体用1L乙腈溶解,再将2L水滴加至体系中,晶体完全析出后搅拌2h,固体用MeCN:水=1:2的混合溶剂洗涤。30-50℃烘干得到固体。收率70.5%。HPLC纯度98.52%。
本实施例所得式5化合物晶型的XRPD、DSC和TGA如图1、图3和图2所示。
实施例3
式5化合物的制备
向5L三口圆底反应烧瓶中依次加入实施例1制备的式3化合物(300g)、式4化合物即2-溴-4氰基-苯乙酮(215g)以及95%乙醇(1.5L),搅拌下升温至油温85℃,内温76℃,升温过程中体系颜色变黄绿色,保温搅拌2h。取样监测,TLC显示M1点消失,新点生成。
降温至20℃以下,搅拌下,将饱和的NaHCO3溶液缓慢的加入反应液中,调节pH值至4-5,再将反应液慢慢倒入纯化水(1.5L)中,有大量白色固体析出,抽滤后滤饼自然晾至半干,晾干时间大约16h。向半干固体中加入DCM(1.2L)和纯化水(600ml),充分搅拌萃取,分液,水相用DCM(150ml)萃取两次,合并有机相,用饱和NaCl溶液洗涤,无水硫酸钠(30g)干燥。过滤去除无水硫酸钠后,向DCM溶液中加入硅胶(200g),室温搅拌吸附2h。过滤去除硅胶,将DCM溶液旋蒸浓缩至淡黄色油状物。将上述油状物溶于EA(300ml)和正庚烷(1.2L),室温搅拌8h析出固体,搅拌初期无固体析出,随着搅拌进行逐渐有白色固体析出,固体有贴壁现象(用刮勺将贴壁固体刮落,继续搅拌)。抽滤,滤饼用EA:正庚烷=1:8的溶液洗涤,抽干滤饼得到白色固体。将白色固体转移至5L三口圆底反应烧瓶中,加入3L异丙醚,室温搅拌24h。抽滤,滤饼用异丙醚洗涤,抽干得到白色固体粉末。
25℃下,将所得固体用0.5L乙醇溶解,再将1L水滴加至体系中,晶体完全析出后搅拌2h,固体用乙醇:水=1:2的混合溶剂洗涤。30-50℃烘干得到330g固体。收率79.5%。HPLC纯度99.52%。
此实施例所得式5化合物晶型的XRPD、DSC和TGA图谱与实施例2所得的晶型XRPD、DSC和TGA图谱一致。
实施例4
式5化合物的制备
向5L三口圆底反应烧瓶中依次加入实施例1制备的式3化合物(300g)、式4化合物即2-溴-4氰基-苯乙酮(215g)以及95%乙醇(1.5L),搅拌下升温至油温85℃,内温76℃,升温过程中体系颜色变黄绿色,保温搅拌2h。取样监测,TLC显示M1点消失,新点生成。
降温至20℃以下,将饱和的NaHCO3溶液缓慢的加入反应液中,调节pH值至4-5,搅拌下,将反应液慢慢倒入纯化水(1.5L)中,有大量白色固体析出,抽滤后滤饼自然晾至半干,晾干时间大约16h。向半干固体中加入DCM(1.2L)和纯化水(600ml),充分搅拌萃取,分液,水相用DCM(150ml)萃取两次,合并有机相,用饱和NaCl溶液洗涤,无水硫酸钠(30g)干燥。过滤去除无水硫酸钠后,向DCM溶液中加入硅胶(200g),室温搅拌吸附2h。过滤去除硅胶,将DCM溶液旋蒸浓缩至淡黄色油状物。将上述油状物溶于EA(300ml)和正庚烷(1.2L),室温搅拌8h析出固体,搅拌初期无固体析出,随着搅拌进行逐渐有白色固体析出,固体有贴壁现象(用刮勺将贴壁固体刮落,继续搅拌)。抽滤,滤饼用EA:正庚烷=1:8的溶液洗涤,抽干滤饼得到白色固体。将白色固体转移至5L三口圆底反应烧瓶中,加入3L异丙醚,室温搅拌24h。抽滤,滤饼用异丙醚洗涤,抽干得到白色固体粉末。
25℃下,将所得固体用1L乙腈溶解,再将2L水滴加至体系中,晶体完全析出后搅拌2h,固体用乙腈:水=1:2的混合溶剂洗涤。30-50℃烘干得到330g固体。收率75.5%。HPLC纯度98.32%。
此实施例所得式5化合物晶型的XRPD、DSC和TGA图谱与实施例2所得的晶型XRPD、DSC和TGA图谱一致。
- 还没有人留言评论。精彩留言会获得点赞!