基于金属铱配合物的多肽荧光环化方法及环化多肽与流程
本申请是2014年3月6日提交的专利申请号为201410079377.0、名称为“基于金属铱配合物的多肽荧光环化方法及环化多肽”的发明专利申请的分案申请,并要求专利申请号为201310077634.2、专利申请日为2013年3月12日、名称为“一种基于组氨酸与金属铱配合物结合的多肽环化方法”的中国发明专利申请的国内优先权。
本发明涉及一种新型多肽荧光环化方法及环化多肽,特别是一种基于一类与多肽中可选择位点的两个组氨酸结合金属铱配合物,完成两个组氨酸之间多肽序列的环化,同时生成荧光基团,而实现的基于组氨酸与金属铱配合物结合的多肽荧光环化方法。
背景技术:
近年来,多肽和各种小分子蛋白由于具有优越的生物相容性和可塑性,被广泛的应用于多种疾病的治疗。作为细胞内大部分信号通路的执行者,这些关键的多肽分子可以通过调控与疾病相关的信号通路,提供治疗药物的作用靶点。此外,作为生物活性分子,多肽来源广泛,易于合成,而且不同的多肽可以提供穿膜,靶向,治疗等多种功能,日益成为当代医学与生命科学研究领域的重要工具之一。
环化多肽在自然界中分布广泛,植物、动物、低等海洋生物、微生物、细菌和病菌等都含有微量的环化多肽。这些环肽的含量虽然低,但其中不少具有明显的生理活性,也因此受到国内外许多化学家、生物学家和药学家的重视。肽的环化因此成为一种重要的多肽分子设计方法,环化能降低环组分的自由度,并稳定多肽的特定二级结构。研究表明,限制多肽的构象能够增加与受体结合的亲和力和选择性,其环化状态下的各项性能均明显优于其线性状态。例如,rgd肽(含有精氨酸-甘氨酸-天冬氨酸的三肽序列)可与多种整合素特异性结合,能有效地促进细胞对生物材料的粘附,达到靶向整合素高表达细胞的目的。研究表明,其环化以后,在稳定性,靶向性以及亲和力各方面都明显优于线性的rgd。
传统的线性多肽环化方法有2种:一类是通过氨基和羧基的缩合,形成肽键。这种传统的首尾环合方法涉及部分或全部的保护和脱保护策略并需要缩合试剂进行相关环反应,过程复杂,且环化后难以做结构的延伸,无法做更复杂的设计与功能化。另一类通过引入二硫键(s-s)对生物活性肽进行环化。通常先合成带有巯基保护的半胱氨酸(cys)残基的线型肽前体,然后脱去cys的保护基,氧化成二硫键后成环,或直接将带保护基的cys氧化成为s-s键,但是s-s键在天然的生物体系的还原环境下不稳定,大大限制了这类生物活性环化多肽的进一步应用。
技术实现要素:
本发明的主要目的在于提供一种环化多肽及基于金属铱配合物的多肽荧光环化方法,其工艺简单、快捷、易于实施,且所获产物具有低毒性,高稳定性等特点,从而克服了现有技术中的不足。
为实现上述发明目的,本发明采用了如下技术方案:
一种环化多肽,包含主要由环金属铱配位化合物与分布在多肽肽链中的不同位置处的两个组氨酸残基通过配位反应而形成的环化结构。
一种基于组氨酸与金属铱配合物结合的多肽环化方法,包括:
提供环金属铱配位化合物,并使所述环金属铱配位化合物与分布在多肽肽链中的不同位置处的两个组氨酸残基发生配位反应,实现多肽环化,并产生可由紫外光激发的绿色荧光,其中,所述多肽肽链的n端不含组氨酸残基或含有被保护的组氨酸残基。
具体而言,所述环金属铱配位化合物的化学分子式为(lc^n)2ir(lsolv)2m,其结构式如下:
其中,lc^n为c^n二齿配体,lsolv为溶剂分子。
进一步的,所述c^n二齿配体可选用但不限于2-苯基吡啶,1-苯基异喹啉,2-苯基苯并噻吩,苯并喹啉,4-甲基苯基吡啶,4,6-二氟苯基吡啶,2-(苯并噻吩)吡啶,2-苯基喹啉和2,5-二苯基恶唑。
进一步的,所述溶剂分子可选自但不限于水、乙腈、dmf或dmso等。
进一步的,m-可选自但不限于三氟甲基磺酸根(otf-)、六氟磷酸根(pf6-)、四氟硼酸根(bf4-)。
进一步的,所述配位反应是在液相环境中进行,所述液相环境主要由缓冲液体系组成,所述缓冲液体系可选用但不限于磷酸缓冲液(pbs)、tris缓冲液、bis-tris缓冲液、mes缓冲液或hepes缓冲液。
进一步的,在所述多肽肽链内,与所述环金属铱配位化合物发生配位反应的两个组氨酸之间间隔0-5个除组氨酸之外的任一种氨基酸,尤其优选间隔2-3个除组氨酸之外的任一种氨基酸。
进一步的,所述多肽可选用但不限于rgd肽等。
与现有技术相比,本发明的优点至少在于:
(1)本发明提供了一种新颖,简便,稳定的多肽环化方式,其通过将环金属铱配位化合物与多肽肽链中的2个组氨酸残基(histidine)结合,使组氨酸之间的多肽序列环化,同时实现荧光标记,并且,即便多肽的序列不同,只要肽链中含有2个组氨酸残基即可以完成其间多肽序列的环化,反应过程快速、简便,反应条件温和,且环化产物稳定、不可逆,在生物体系内无毒性,便于继续进行复杂的修饰和组装过程,可被广泛应用于分析检测、荧光成像等技术领域。
(2)本发明的环化反应可在常温下、常用的缓冲溶液中进行,不需要额外的交联试剂,反应条件温和,操作步骤简单,反应过程快速,反应效率高,成本低廉;
(3)本发明所使用的环金属铱配位化合物在与多肽的组氨酸残基结合之前不具备荧光,配位过程中形成的复合物才有明显的荧光增强,从而一步完成结构的优化(环化)以及荧光的标记,同时产生的荧光结合化学表征手段(质谱,uplc)可以准确的证明多肽环化的成功与否,并且,本发明的环金属铱配位化合物还具有较好的光稳定性,较长的荧光寿命等光物理学特性和化学稳定性,在常规条件下以固体或溶液形式可以长时间保存。
(4)本发明在实际的应用过程中,在赋予多肽(如,rgd)环化的结构(ir-hrgdh,此处的ir系环金属铱配位化合物的简写)之后,还可体现出与线性状态(rgdhh-ir,此处的ir含义同上)相比更好的生物选择性,表明本发明方法可以达到传统环化手段所完成的优化多肽结构与功能的效果。
附图说明
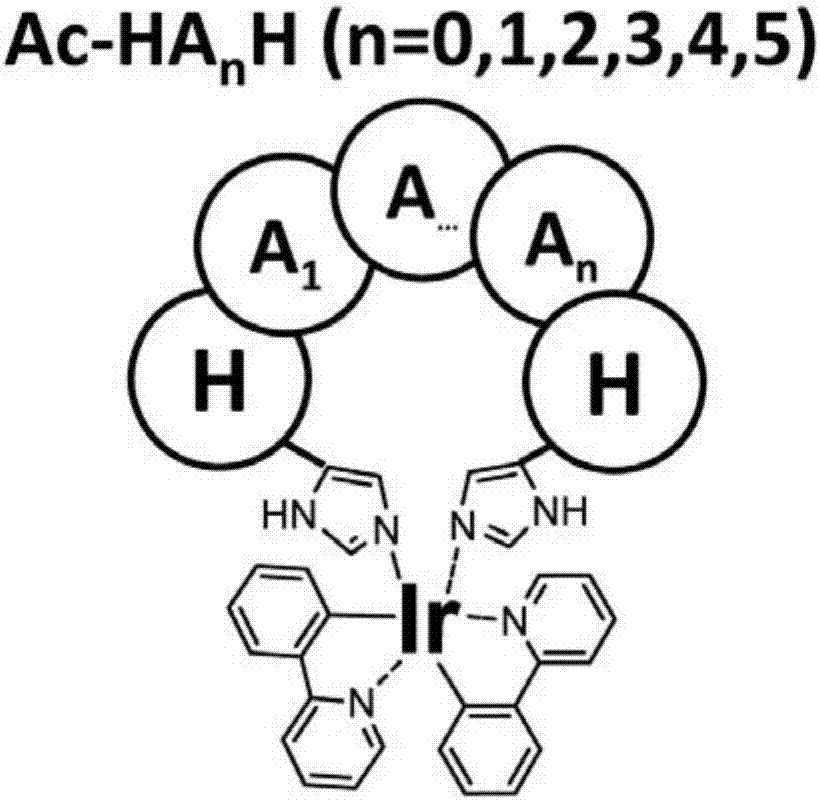
图1是本发明一较为优选的实施方案中一种基于金属铱配合物的环化多肽的结构示意图,其中,h为组氨酸,x为氨基酸,n为大于或等于零的整数;
图2a-图2d分别展示了实施例1中利用模式多肽ac-hanh-nh2(简写为“hanh”)(n=0,1,2,3,4,5)与环金属铱配位化合物(简写为“ir”,下同)发生配位反应的产物结构、反应速率,产生的荧光强度以及还原环境下(gsh=5mm)的稳定性图谱;
图3是本发明实施例2-3中多肽ac-hrgdh-nh2(简写为“hrgdh”)或ac-hrgdh-(klaklak)2(简写为“hrgdh-kla”或“hrgdh-(klaklak)2”)与环金属铱配位化合物完成环化后,环化部分的结构示意图,图中虚线表示末端羧基酰胺化或与后续多肽序列连接;
图4是本发明实施例1、2中环金属铱配位化合物、环化多肽ir-hrgdh,ir-hrgdh-(klaklak)2在配位环化前后的荧光发射光谱图(ex:328nm);
图5是本发明实施例2中环金属铱配位化合物与多肽ac-hrgdh-nh2完成配位反应后产物(ir-hrgdh)的质谱;
图6是本发明实施例2中环金属铱配位化合物,多肽ac-hrgdh-nh2以及二者反应产物(ir-hrgdh)的uplc;
图7a-图7c是本发明实施例2中多肽rgd在环化后于不同细胞系(a549、mcf-7)中的选择性入胞的激光共聚焦成像图,其中,由图7a和图7b可以看到,环化后的rgd(ir-hrgdh)与线性的rgd(rgdhh-ir)在入胞能力上的明显差异;
图8是本发明实施例3中ac-hrgdh-(klaklak)2与环金属铱配位化合物完成配位反应前后(即环化前后)的细胞毒性与溶血性变化比较;
图9是本发明实施例3中所获环化多肽ir-hrgdh-(klaklak)2(亦可简称为ir-hrgdh-kla)快速诱发细胞凋亡全过程的共聚焦成像图谱。
具体实施方式
如前所述,鉴于现有技术中的不足,本发明的主旨在于提供一种新型的基于金属铱配合物的多肽荧光环化方法,其主要是基于一类与组氨酸结合生成荧光基团的铱配合物而实现。
进一步的讲,作为本发明的一个较为优选的实施方案,该基于金属铱配合物的多肽荧光环化方法系通过一类自身无荧光的环金属铱配位化合物与多肽肽链上的2个组氨酸残基结合,完成多肽环化,优化多肽性能的同时形成可被激发的荧光基团,得以标记多肽分子,并用于分析检测及荧光成像。
本发明中多肽环化的过程简单、快速、高效,反应条件温和,环化后的多肽性质稳定,无明显生物毒性,可应用于进一步的复杂修饰和组装过程,而且带有绿色荧光便于示踪,在生命科学等领域有着广阔的应用前景,例如,其应用范围可包括但不局限于生物分子检测、细胞染色、动物组织标记和多肽的组装,标记及示踪等实验。
其中,环金属铱配位化合物优选采用由两个相同的c^n二齿配体以及两个溶剂分子配位络合的环金属铱配位化合物,其化学分子式可以为(lc^n)2ir(lsolv)2m-,其结构式可参阅前文内容。
进一步的,前述c^n二齿配体、溶剂分子的类型亦可参阅前文,此处不再赘述。
在一典型实施例中,前述环金属铱配位化合物的结构式可以为:
其中ppy为2-pyridine(2-苯基吡啶),otf-为trifluoromethanesulfonate(三氟甲基磺酸根),lsolv为易于从中心金属脱离的溶剂分子,比如水或乙腈。
前述otf-亦可以六氟磷酸根(pf6-)、四氟硼酸根(bf4-)等替换。
显然的,为实现前述配位反应,则所述多肽肽链内必需至少含有两个组氨酸,其中用以与环金属铱配位化合物进行配位反应的两个组氨酸应分布于多肽肽链上的两个不同选定位置。特别是对于线形多肽而言,该两个组氨酸应沿长度方向分布在多肽肽链的不同位置处。
又例如,前述环金属铱配位化合物与多肽反应的过程可参考图1,其中h为组氨酸残基、x为间隔于两个组氨酸残基之间的、除组氨酸之外的任意氨基酸,其数量可以是大于或等于0的正整数,优选为0-5个,并且分布于间隔区段的氨基酸种类不限(当然,组氨酸除外)。
进一步的,本案发明人发现,受空间位阻影响,多肽肽链内两个选定组氨酸残基之间间隔的氨基酸数量为2-3个时,环化效果(包括反应速率,产生荧光强度,产物稳定性等)最佳。
又及,前述多肽肽链的n端应不含组氨酸残基或含有被保护的组氨酸残基。更具体的讲,在该方法中,环金属铱配位化合物在进行配位反应时,需同时提供2个配位基团,例如咪唑,氨基等。因此必需由多肽肽链中的两个组氨酸分别提供一个自由的咪唑基团,或多肽n端单个组氨酸同时提供咪唑基团与末端氨基,方可完成该配位反应。因此,为避免可能存在于多肽肽链n端的、且保留氨基的组氨酸残基同时提供咪唑基团和末端氨基参与与环金属铱配位化合物的配位络合反应,应将该n端的组氨酸氨基进行保护(如乙酰化)。
而若一个组氨酸位于多肽c端或内部,则其均无法单独完成前述配位反应,也无法快速的产生明显的绿色荧光,更无法实现多肽环化。
以下结合若干较佳实施例对本发明的技术方案作进一步的说明。
实施例1:模式多肽ac-hanh-nh2(n=0,1,2,3,4,5)的环化效果比较
将[(ppy)2ir(h2o)2](otf)与首尾各带一个组氨酸的模式多肽ac-hanh(n=0,1,2,3,4,5)反应(前述a代表丙氨酸),[(ppy)2ir(h2o)2](otf)与多肽的反应浓度的摩尔比为1:1,反应在pbs(ph7.4)中进行,37℃震荡2小时或4℃过夜,从而获得环化后的多肽。
在紫外光(365nm)照射下,环化后的多肽可以产生绿色荧光。首尾组氨酸中间间隔氨基酸数量不同,配位反应的速率,生成荧光的强度以及在模拟细胞浆内还原条件(gsh=5mm)下的稳定性略有不同。(参阅图2a-图2d)分析比较两个组氨酸之间间隔氨基酸数量变化发现,间隔氨基酸数量在0-5个之间时,间隔2或3个氨基酸的环化肽在反应速率,产生荧光强度和还原条件下稳定性方面都较间隔0,1,4,5个氨基酸的环化多肽有明显优势。其中,当n=0,1时,间隔氨基酸过少,导致两个组氨酸间隔短,柔性不足,反应受空间位阻的影响;当n=4,5,则间隔氨基酸又过多,两个组氨酸距离过远,同样限制的反应的进行。当两个组氨酸之间间隔2-3个氨基酸时,为最佳选择。
实施例2:多肽ac-hrgdh-nh2的环化反应
将[(ppy)2ir(h2o)2](otf)与首尾各带一个组氨酸的多肽ac-hrgdh-nh2反应(参阅图3),多肽环化过程及反应条件与实施例1基本相同,不同之处在于本实施例中的多肽为ac-hrgdh-nh2。图4显示了铱配位化合物(图中简写为ir)与多肽ac-hrgdh-nh2(图中简写为hrgdh)配位环化前后的荧光发射光谱(ex:328nm)。并且,请参阅图5,以质谱对反应产物进行分析,所得测试数值(1162.4)与预估反应产物(ir-hrgdh)分子量(1163)相符。
再请参阅图6,经uplc(超高效液相色谱)测试显示,经过反应后,原本属于环金属铱配位化合物(图中简写为ir)和多肽(图中简写为ac-hrgdh)的特征峰完全消失,并生成一个新峰(ir-hrgdh),反应完全程度大于95%。
再请参阅图7a-7c,可以看到,利用本实施例的方法完成前述rgd多肽序列的环化后,体现出了较其线性状态明显优化的入胞能力和在rgd识别阳性(a549)/阴性(mcf-7)细胞间的选择性。这与文献报道的其他传统环化方式对rgd产生的优化作用一致。并且,本实施例中环化后多肽的mtt检测结果表明通过此类环化方法完成环化造成的多肽细胞毒性低,lc50大于200μm,其结果请参阅表1所示。
实施例3:多肽ac-hrgdh-(klaklak)2的环化与细胞毒性优化
本实施例中,多肽环化部分结构(亦可参考图3)与环化过程及反应条件与实施例2基本相同,不同之处在于,本实施例中的多肽在环化部分之外连接了有细胞毒性的多肽(klaklak)2。(klaklak)2本身在单体情况下因入胞困难,无法进入细胞诱发细胞凋亡。而通过与ir-hrgdh相连接,促进了其与rgd识别呈阳性的细胞表面的粘附,有效的帮助其入胞并发挥毒性。
ac-hrgdh-(klaklak)2与环金属铱配位化合物完成配位环化反应前后的细胞毒性与溶血性变化比较如图8所示。与环金属铱配位化合物完成配位环化反应后,多肽体现出了毒性40倍的提高,而溶血性质仍然保持在较低的水平,表面其具有极佳的活体应用前景。
本实施例中相关的荧光发射光谱结果如图2所示,细胞毒性mtt检测结果如下表1所示。
表1实施例1-3中所涉及的多肽及环化多肽的细胞毒性lc50数据。
同时,由于本发明在完成多肽环化的同时也为环化多肽标记上绿色荧光,使本实施例中环化后的多肽ir-hrgdh-(klaklak)2引发细胞凋亡的进程得以表征(参阅图9)。
本发明利用自身无荧光的环金属铱配位化合物与肽链中选定位置的2个组氨酸发生配位反应,完成多肽环化,同时形成可被激发的荧光基团,使多肽分子被标记荧光,可用于分析检测及荧光成像等。
总之,本发明工艺简单快速,反应条件温和,反应效率高,产物性能稳定,无明显生物毒性,利于进一步复杂的设计修饰或组装,具有广阔生物化学和医学应用前景。
需要指出的是,本发明所揭示的乃较佳实施例的一种或多种,凡是局部的变更或修饰而源于本发明的技术思想而为熟习该项技术的人所易于推知的,俱不脱离本发明的专利权范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种羰基锝-99m标记的美法仑配合物及其制备方法与用图
- 一种2-羰基-3-苯基丙酸苯甲酰腙二苯基锡配合物及其制备方法和应用
- 一种新型金属有机配合物纤维及其衍生多孔碳纤维的制备方法
- 包含新的配体结构的金属配合物的制作方法
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用
- 一种2-羰基-2-苯基乙酸苯甲酰腙二丁基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸水杨酰腙二苯基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用
- 一种2-羰基-2-苯基乙酸苯甲酰腙二丁基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸水杨酰腙二苯基锡配合物及其制备方法和应用