检测靶核酸序列的引物、荧光探针及方法与流程
本发明属于基因工程技术领域,更具体地,本发明涉及一种检测靶核酸序列的引物、荧光探针及方法。
背景技术:
荧光pcr技术是检测靶核酸序列的常用技术,目前常用的荧光pcr检测方法包括taqman探针法、分子信标(molecularbeacon)探针法、荧光杂交双探针(hybridizationprobe)法等。其中荧光杂交双探针法主要通过使用两条可与靶序列同一条单链杂交的荧光探针,其中一条探针3’端修饰一种荧光基团,另一条探针5’端修饰另一种荧光基团,当靶序列存在时通过非对称pcr产生大量可与这两条荧光探针反向互补的单链,当两条荧光探针首尾相连与反向互补的单链dna杂交时,第一条探针3’端的荧光基团与第二条探针5’端的另一个荧光基团在空间距离上缩短,引发荧光能量共振转移,所引起的荧光信号强弱变化可以用来检测靶序列的存在。
荧光探针溶解曲线分析技术是基于不对称pcr扩增技术的一种定性检测方法。利用分子信标探针、fret探针或taqman探针在不对称pcr反应后进行溶解曲线分析,可因为所结合序列变化所造成的溶解曲线峰值变化,从而分辨不同的靶序列位点。这项技术主要优势在于可以利用不同荧光通道和不同荧光探针溶解峰温度的组合同时检测多个检测位点,而且设计良好的探针可以通过溶解峰的变化分辨不同的突变位点。
目前荧光探针溶解曲线技术在同一荧光通道使用多条荧光探针进行检测时,即使荧光探针未与靶序列结合,探针自身或探针之间在低温条件下互相缠绕误杂交也会产生背景信号,多条荧光探针的背景信号之间互相干扰叠加,造成荧光背景过高,从而影响了溶解曲线的判读。
技术实现要素:
为了克服上述现有技术的缺陷,本发明提供了一种检测靶核酸序列的引物、荧光探针及方法,该方法基于引物上淬灭基团对杂交探针荧光进行淬灭,使得多条检测探针的背景信号之间不会互相干扰叠加。
为了实现上述发明目的,本发明采取了以下技术方案:
一种检测靶核酸序列的引物,该引物的近3’端修饰有淬灭基团或荧光基团。
在其中一些实施例中,所述淬灭基团为bhq1、bhq2、或bhq3,所述荧光基团为fam、hex、rox、或cy5。
在其中一些实施例中,所述引物为正向引物或反向引物。
在其中一些实施例中,所述淬灭基团或荧光基团与所述引物的3’端末尾距离1-20个碱基。
在其中一些实施例中,所述淬灭基团或荧光基团与所述引物的3’端末尾距离1-10个碱基。
本发明还提供了一种检测靶核酸序列的荧光探针,所述荧光探针与权利要求1~5任一项所述的引物与dna的不同单链互补,所述荧光探针的近3’端修饰有荧光基团或淬灭基团,且当所述引物修饰有荧光基团时,所述荧光探针修饰有淬灭基团,当所述引物修饰有淬灭基团时,所述荧光探针修饰有荧光基团。
在其中一些实施例中,所述淬灭基团为bhq1、bhq2、或bhq3,所述荧光基团为fam、hex、rox、或cy5。
在其中一些实施例中,所述荧光基团或淬灭基团与荧光探针的3’端末尾距离0-20个碱基。
在其中一些实施例中,所述荧光基团或淬灭基团与荧光探针的3’端末尾距离0个碱基,即直接修饰在3’端末尾。
本发明还提供了一种检测靶核酸序列的方法,包括以下步骤:
以待检测核酸序列为模板,以上述引物及荧光探针进行pcr反应,反应后进行溶解曲线分析,绘制溶解曲线,即可。
本发明的检测靶核酸序列的荧光探针3’端修饰荧光基团,扩增靶序列的引物或通用引物3’端修饰淬灭基团。当检测靶序列的pcr反应未进行时,荧光探针上的荧光基团处于正常状态,可检测到高强度的荧光信号。当检测靶序列的pcr反应进行后,荧光探针与pcr反应过程产生的大量单链pcr产物结合,所发出的荧光被淬灭引物或通用淬灭引物上的淬灭基团淬灭,使荧光强度下降。当进行溶解曲线分析时,荧光探针在特定温度下与所结合的单链pcr产物分离,所发出的荧光不再被淬灭引物或通用淬灭引物上的淬灭基团淬灭,从而荧光强度上升,形成特定温度处的反向溶解峰。
当荧光基团修饰在引物或通用引物的3’端时,检测靶核酸序列的探针3’端修饰淬灭基团。当检测靶序列的pcr反应未进行时,引物上的荧光基团处于正常状态,可检测到高强度的荧光信号。当检测靶序列的pcr反应进行后,修饰有淬灭基团的探针与pcr反应过程产生的大量单链pcr产物结合,引物上修饰的荧光基团发出的荧光被探针上的淬灭基团淬灭,使荧光强度下降。当进行溶解曲线分析时,探针在特定温度下与所结合的单链pcr产物分离,引物上荧光基团所发出的荧光不再被探针的淬灭基团淬灭,从而荧光强度上升,形成特定温度处的反向溶解峰。
与现有技术相比,本发明具有以下有益效果:
本发明的基于引物近3’端修饰的淬灭基团淬灭特异性荧光探针3’端荧光基团,或基于荧光探针近3’端修饰的淬灭基团淬灭特异性引物3’端荧光基团,并进行溶解曲线分析方法与其它溶解曲线分析技术相比,当检测靶序列不存在情况下,荧光探针产生的荧光信号不受淬灭基团影响,荧光强度基本保持不变,使得杂交曲线分析时的背景本底明显降低,不会影响其它阳性靶序列的检测;当有多条荧光探针存在时,由于未结合的荧光探针在温度变化时三维结构的改变不会像普通taqman探针或分子信标探针一样明显改变荧光信号强度,荧光探针之间的互相缠绕也不会像普通taqman探针或分子信标探针一样互相改变荧光信号强度,使得探针之间的本底信号不会互相干扰,使得多重pcr的判读更加清晰。
附图说明
图1为本发明实施例1的检测靶核酸序列的引物及荧光探针的结构示意图;
图2为本发明实施例1的检测靶核酸序列的通用引物及荧光探针的结构示意图;
图3为本发明实施例1的基于淬灭基团的引物和荧光探针杂交分析检测方法的原理图;
图4为本发明实施例2的检测结果图;
图5为本发明实施例2的检测结果图。
具体实施方式
下面结合附图和具体实施例进一步叙述本发明,本发明未述及之处适用于现有技术。下面给出本发明的具体实施例,但实施例仅是为了进一步详细叙述本发明,并不限制本发明的权利要求。
实施例1基于荧光能量共振转移(fret)的检测靶核酸序列的方法
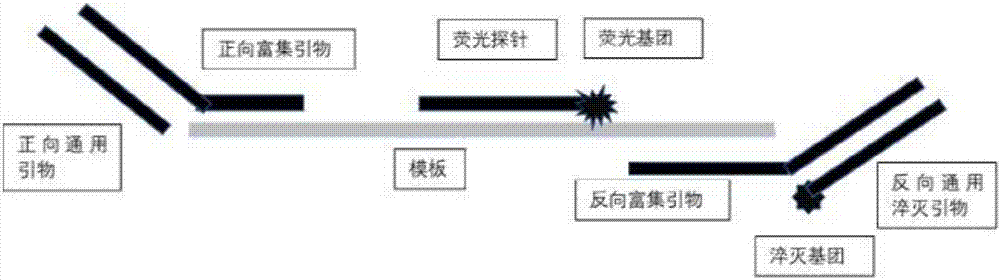
请参阅图1~2,为本发明的检测靶核酸序列的引物或通用引物及荧光探针的结构示意图。
引物可设计为正向引物或反向引物,本实施例中为反向引物,近3’端修饰有淬灭基团bhq1。荧光探针与引物与dna的不同单链互补,近3’端修饰有荧光基团fam,该荧光基团可被引物上的淬灭基团淬灭,使得荧光强度下降(图1)。
正向和反向富集引物的3’端与模板反向互补,5’端为通用引物结合部位。通用引物可设计为正向引物或反向引物,本实施例中为反向,近3’端修饰有淬灭基团bhq2。荧光探针与通用引物对应的富集引物与dna的不同单链互补,近3’端修饰有荧光基团rox,该荧光基团可被通用引物上的淬灭基团上淬灭,使得荧光强度下降(图2)。
请参阅图3a~c,为本发明的基于淬灭基团的引物和荧光探针杂交分析检测方法的原理图;
在待检测模板不存在情况下,荧光探针、通用引物均处于游离状态,荧光基团与淬灭基团距离较远,不发生fret效应,荧光基团所发出的荧光强度保持基本不变(图3a)。
当待检测模板存在的情况下,引物与待检测模板结合,进行不对称pcr反应,产生大量由引物延伸形成的单链pcr产物(图3b)。
荧光探针与引物延伸形成的单链pcr产物互补结合,使得荧光基团与淬灭
基团相互靠近,发生fret现象,检测到的荧光强度明显下降。在进行溶解曲
线分析时,随着温度升高,荧光探针从结合的pcr产物上分离,fret现象消
失,荧光强度重新上升(图3c)。
实施例2使用淬灭基团修饰的引物及荧光基团修饰的探针检测不同浓度靶序列模板
根据本发明实施例1的原理,采用本实施例的方法检测不同浓度靶序列模板,以评价其实用性。
靶序列模板为包含pcr扩增区域的人工质粒,由生工科技(上海)有限公司合成,按浓度稀释为103/ul、104/ul不同浓度。
使用仪器为上海宏石slan-96p荧光定量pcr仪。
引物序列为:
std-f:tcagcaactagcttaccatc(seqidno:1),3’端倒数第2位t修饰bhq1淬灭基团
std-r:gctgacgttgcaagaagacg(seqidno:2)
荧光探针序列为:
std-p:tgatcatgtaatggaaggggcaaga(seqidno:3),3’端修饰hex荧光基团。
反应体系为:
反应条件:
熔解曲线分析:
设置检测荧光通道为hex
反应条件为:95℃2min;45℃2min;45℃-85℃熔解曲线分析。
结果如图3所示,从图4可知,以103/ul、104/ul浓度质粒各1ul作为模板,反应后溶解曲线分析在63℃左右有明显的倒置溶解峰,空白对照孔无对应的倒置溶解峰。
实施例3使用荧光基团修饰的引物及淬灭基团修饰的探针检测不同浓度靶序列模板
根据本发明实施例1的原理,采用本实施例的方法检测不同浓度靶序列模板,以评价其实用性。
靶序列模板为包含pcr扩增区域的人工质粒,由生工科技(上海)有限公司合成,按浓度稀释为103/ul、104/ul不同浓度。
使用仪器为上海宏石slan-96p荧光定量pcr仪。
引物序列为:
std-f:tcagcaactagcttaccatc(seqidno:1),3’端倒数第2位t
修饰fam荧光基团
std-r:gctgacgttgcaagaagacg(seqidno:2)
荧光探针序列为:
std-p:tgtaatggaaggggcaaga(seqidno:3),3’端修饰bhq1淬灭基团。
反应体系为:
反应条件:
熔解曲线分析:
设置检测荧光通道为fam
反应条件为:95℃2min;45℃2min;45℃-85℃熔解曲线分析。
结果如图5所示,从图5可知,以103/ul、104/ul浓度质粒各1ul作为模板,反应后溶解曲线分析在59℃左右有明显的倒置溶解峰,空白对照孔无对应的倒置溶解峰。
试验例1本发明的检测方法的敏感性试验
将淋球菌(ng)的质粒标准品分别做8个稀释度从1×107~1×100拷贝数
/μl,以倍比稀释为模板分别按实施例2的方法进行荧光定量pcr扩增,存在tm值63±1摄氏度的倒置溶解峰判读为阳性,否则判读为阴性。同时利用引物对seqidno:1和seqidno:2进行常规pcr扩增。
检测结果表明,利用本发明的方法检测ng标准质粒的灵敏度可达1×102拷贝数/μl,而普通pcr最低可以检测到1×102拷贝数/μl,说明本发明的检测方法与普通pcr的检测方法相同。
试验例2本发明的检测方法的特异性试验
通过本发明实施例2的检测方法,分别对国家标准品菌种样本:淋球菌、人乳头瘤病毒、沙眼衣原体、淋球菌、微小脲原体、阴道毛滴虫、生殖道支原体、人型支原体进行检测,结果表明,除淋球菌标准品会有阳性溶解峰外,其他病原体样本均没有阳性溶解峰,证明本发明建立的检测方法特异性强,与其他常见的生殖道病原体均无交叉反应。
试验例3本发明的检测方法的重复性试验
以ng质粒标准品中的1×106和1×104拷贝数/μl两个稀释度的作为模板,对这两个稀释度在不同时间段进行2次重复测定,每次对同一模板同时进行3次重复测定及设置3个重复孔,按照本发明实施例2所述的方法进行实时荧光定量pcr。其中:变异系数(β)=标准偏差(sd)/平均数(x),结果重复性试验阳性率100%。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
序列表
<110>江西贤聚景欣医药生物科技有限公司
<120>检测靶核酸序列的引物、荧光探针及方法
<130>3
<170>patentinversion3.1
<210>1
<211>20
<212>dna
<213>人工序列
<400>1
tcagcaactagcttaccatc20
<210>2
<211>20
<212>dna
<213>人工序列
<400>2
gctgacgttgcaagaagacg20
<210>3
<211>25
<212>dna
<213>人工序列
<400>3
tgatcatgtaatggaaggggcaaga25
- 还没有人留言评论。精彩留言会获得点赞!
- 用于检测AEDs引起的SJS/TEN的引物、试剂盒及方法与制造工艺
- 检测植原体的LAMP引物组及其试剂盒和应用的制造方法与工艺
- 用于多重PCR检测致腹泻寄生虫的引物组及试剂盒的制造方法与工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 一种基于特异性SS-COI引物的丽草蛉PCR快速检测方法与制造工艺
- 一种利用线粒体D-loop区特异区域鉴定鼋的标记引物及方法
- 一种用于检测大豆拟茎点种腐病菌的特异性lamp引物组合物及其应用
- 一种针对膨胀污泥中Eikelboom Type 0092型丝状菌群特异性引物和方法
- 紫苑石斛的分子特异性标记引物及检测方法
- 一种检测牛hcd携带者的特异性pcr引物、试剂盒及检测方法
- 检测植原体的LAMP引物组及其试剂盒和应用的制造方法与工艺
- 用于多重PCR检测致腹泻寄生虫的引物组及试剂盒的制造方法与工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 一种利用线粒体D-loop区特异区域鉴定鼋的标记引物及方法
- 一种用于检测大豆拟茎点种腐病菌的特异性lamp引物组合物及其应用
- 一种针对膨胀污泥中Eikelboom Type 0092型丝状菌群特异性引物和方法
- 紫苑石斛的分子特异性标记引物及检测方法
- 一种检测牛hcd携带者的特异性pcr引物、试剂盒及检测方法
- 用于检测牛源性成分的lamp引物及其设计方法
- 七种不同种属来源细胞的pcr检测引物和检测方法与应用
- 检测植原体的LAMP引物组及其试剂盒和应用的制造方法与工艺
- 用于多重PCR检测致腹泻寄生虫的引物组及试剂盒的制造方法与工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 一种利用线粒体D-loop区特异区域鉴定鼋的标记引物及方法
- 一种用于检测大豆拟茎点种腐病菌的特异性lamp引物组合物及其应用
- 一种针对膨胀污泥中Eikelboom Type 0092型丝状菌群特异性引物和方法
- 紫苑石斛的分子特异性标记引物及检测方法
- 一种检测牛hcd携带者的特异性pcr引物、试剂盒及检测方法
- 用于检测牛源性成分的lamp引物及其设计方法
- 七种不同种属来源细胞的pcr检测引物和检测方法与应用