苯甲酸阿格列汀杂质的制备方法与流程
本发明涉及一种苯甲酸阿格列汀杂质的制备方法,属于医药化工技术领域。
背景技术:
苯甲酸阿格列汀(alogliptinbenzoate),化学名为:2-[[6-[(3r)-3-氨基-1-哌啶基]-3,4-二氢-3-甲基-2,4-二氧代-1(2h)-嘧啶基]甲基]苯甲腈苯甲酸盐,化学结构如下:
苯甲酸阿格列汀是日本takeda公司研发的丝氨酸蛋白酶二肽基肽酶iv(dpp-iv)抑制剂,具有高选择性,能维持体内胰高血糖素样肽1(glp-1)和葡萄糖依赖性促胰岛素多肽(gip)的水平,促进胰岛素的分泌,从而发挥降糖疗效,2010年4月获得日本厚生劳动省的上市批准,是目前我国已批准的第5个dpp-4抑制剂,用于2型糖尿病患者的血糖控制。
现有文献和资料均有报道苯甲酸阿格列汀的合成路线以及苯甲酸阿格列汀相关杂质,主要有:2-[[6-[(3s)-3-氨基-1-哌啶基]-3,4-二氢-3-甲基-2,4-二氧代-1(2h)-嘧啶基]甲基]苯甲腈苯甲酸盐、(r)-n,1-二(1-(2-氰基苯甲基)-3-甲基-2,4-二氧代嘧啶-6-基)-3-氨基哌啶、2-((3-甲基-2,4,6-三氧代-四氢嘧啶-1-基)甲基)苯腈等,但尚未见本发明所公开杂质。
技术实现要素:
本发明的目的在于提供一种苯甲酸阿格列汀杂质的制备方法,其操作便捷,反应条件温和可控,降低了副反应的发生,使得目标产物苯甲酸阿格列汀易于分离纯化,纯度高。
本发明所述的苯甲酸阿格列汀杂质的制备方法,包括以下步骤:
(1)以6-氯-3-甲基嘧啶-2,4(1h,3h)-二酮为原料,与2-溴甲基苯腈反应,制得tm12-((6-氯-3-甲基-2,4-二氧代-3,4-二氢嘧啶-1(2h)-基)甲基)苯腈;
(2)tm1在碱性条件下与一元醇类反应,制得苯甲酸阿格列汀杂质bp1;bp1的结构式如下:
反应方程式如下:
步骤(1)中,反应在二异丙基乙胺存在的条件下进行,反应所用溶剂为甲苯和n-甲基吡咯烷酮。n-甲基吡咯烷酮与甲苯的体积比为0.8-1.2:1。
所述二异丙基乙胺与2-溴甲基苯腈的摩尔比为1-2:1。
所述2-溴甲基苯腈与6-氯-3-甲基嘧啶-2,4(1h,3h)-二酮的摩尔比为1-1.5:1。
步骤(1)中,反应温度为68-73℃,反应时间为1-3小时。
步骤(2)中,碱性条件为在碳酸氢钠存在的条件下,反应所用的溶剂为水。
步骤(2)中,反应温度为68-73℃,反应时间为8-16小时。
对于苯甲酸阿格列汀杂质bp1的研究是一个动态发展和不断推进的过程,其在目标产物中难以除去,将其应用于苯甲酸阿格列汀杂质分析,控制杂质在苯甲酸阿格列汀的原料或制剂中的含量,即将bp1作为对照品,进一步完善苯甲酸阿格列汀原料药的质量标准。
本发明与现有技术相比,具有以下有益效果:
(1)所述的制备方法操作便捷,反应条件温和可控;
(2)降低了副反应的发生,使得目标产物苯甲酸阿格列汀易于分离纯化,纯度高。
附图说明
图1为所述的苯甲酸阿格列汀杂质bp1的核磁氢谱;
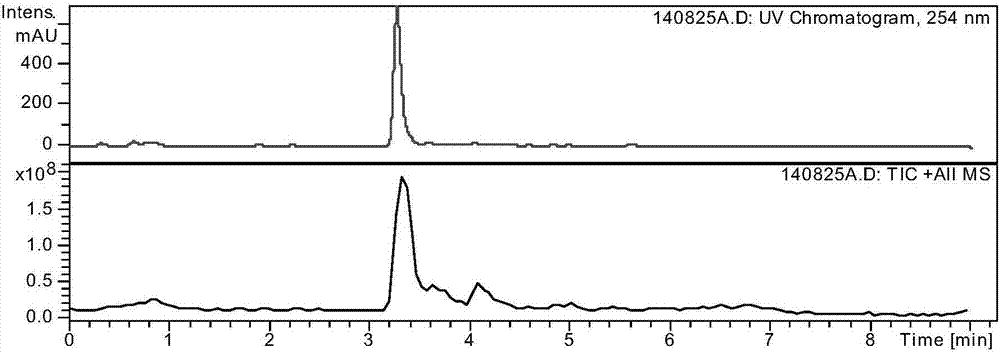
图2为所述的苯甲酸阿格列汀杂质bp1的液相质谱;
图3为所述的苯甲酸阿格列汀杂质bp1的质谱图。
具体实施方式
下面结合实施例对本发明作进一步的说明,但其并不限制本发明的实施。
实施例1
25℃下将10.0g(62.3mmol)sm1、40mln-甲基吡咯烷酮、12.1g(93.6mmol)二异丙基乙胺依次加入到500ml三口瓶中,在搅拌下再向反应瓶中滴加溶有13.4g(68.4mmol)sm2的40ml甲苯溶液,滴加完毕后,升温至70℃,反应1.5小时。反应体系降至室温,向其中加入40ml水,25℃下搅拌30分钟,降温至0℃,搅拌1小时,抽滤。将滤饼用10ml异丙醇淋洗后烘干得到15.5g类白色固体,收率为90.2%。
将5.5g(20mmol,1eq)tm1加入100ml叔丁醇中,室温25℃下滴入5.0g(60mmol,3eq)碳酸氢钠的50ml水溶液,室温70℃下搅拌12小时。
冷水浴10℃下向体系中滴加浓盐酸,调ph=5-6,40℃水浴旋蒸除去叔丁醇,加入100ml水和100ml二氯甲烷,分液,水相用100ml二氯甲烷萃取三次,合并有机相用无水硫酸钠干燥后,40℃水浴旋蒸除去溶剂,得到棕红色油状物。加入20ml二氯甲烷将此油状物溶解,加入80ml异丙醚,-10℃下冷冻,析出固体,过滤40℃鼓风烘干得黄色固体2.7g,含量99.2%,收率44%。
实施例2
tm1的制备同实施例1。
将5.5g(20mmol,1eq)tm1加入100ml异丙醇中,室温25℃下滴入5.0g(60mmol,3eq)碳酸氢钠的50ml水溶液,室温68℃下搅拌16小时。
冷水浴10℃下向体系中滴加浓盐酸,调ph=5-6,40℃水浴旋蒸除去异丙醇,加入100ml水和100ml二氯甲烷,分液,水相用100ml二氯甲烷萃取三次,合并有机相用无水硫酸钠干燥后,40℃水浴旋蒸除去溶剂,得到棕红色油状物。加入20ml二氯甲烷将此油状物溶解,加入80ml异丙醚,-10℃下冷冻,析出固体,过滤40℃鼓风烘干得黄色固体1.4g,含量99.1%,收率23%。
实施例3
tm1的制备同实施例1。
将5.5g(20mmol,1eq)tm1加入100ml乙醇中,室温25℃下滴入5.0g(60mmol,3eq)碳酸氢钠的50ml水溶液,室温73℃下搅拌8小时。
冷水浴10℃下向体系中滴加浓盐酸,调ph=5-6,40℃水浴旋蒸除去乙醇,加入100ml水和100ml二氯甲烷,分液,水相用100ml二氯甲烷萃取三次,合并有机相用无水硫酸钠干燥后,40℃水浴旋蒸除去溶剂,得到棕红色油状物。加入20ml二氯甲烷将此油状物溶解,加入80ml异丙醚,-10℃下冷冻,析出固体,过滤40℃鼓风烘干得黄色固体2.1g,含量98.8%,收率34.5%。
- 还没有人留言评论。精彩留言会获得点赞!