一种壳聚糖基铅离子印迹纳米纤维的制备方法与流程
本发明涉及一种壳聚糖基铅离子印迹纳米纤维的制备方法,属于吸附材料技术领域。
背景技术:
重金属离子具有极强的毒性,且不可生物降解,通过生物链的方式在人体内聚集,对人体造成不可逆的健康危害。重金属在世界各国均被列为第一优先级控制污染物,去除废水中重金属已成为一个刻不容缓的世界性课题。铅离子作为常见的有毒重金属之一,其主要来源于工业排放的废水,如铅酸电池、燃料电池和涂料等领域。目前铅离子的去除主要通过吸附法、生物吸附、离子交换、膜过滤、化学沉淀和电化学还原等技术。在众多技术中,吸附法是最经济、最有效的处理含铅废水。
壳聚糖作为甲壳素脱乙酰化产物,是自然界中产量仅次于纤维素的天然高分子材料。其无毒,具有良好的生物降解性、生物相容性和生物活性等优点。壳聚糖分子链上的氨基和羧基能与重金属离子配位形成螯合物,因此壳聚糖作为吸附剂被广泛应用于工业重金属废水的处理。然而壳聚糖在水溶液中易于溶胀、酸性条件下溶解和机械性能差等缺点,限制其应用。为了克服以上缺点,常采用环氧氯丙烷、乙二醇缩水甘油酯和戊二醛等交联剂交联。由于壳聚糖上的部分活性点氨基和羧基与交联剂反应失去反应活性,使壳聚糖的吸附容量大大降低。因此交联的壳聚糖必须与含有氨基、羧基和巯基等活性基团接枝反应,以提高吸附容量。
利用离子印迹技术,以不同的金属离子为模板,通过与壳聚糖配位形成螯合金属交联壳聚糖,除去模板后得到离子印迹壳聚糖。以金属离子为模板制得的离子印迹聚合物内部分布有模板分子的印迹孔穴,这些孔穴与模板离子的尺寸大小、空间结构、结合位点分布等方面高度匹配,因而对模板离子具有特意的识别能力,赋予对重金属离子选择性分离能力。然而目前大部分壳聚糖都是以粉末颗粒形成存在,比表面积小、孔隙率低,且难以回收二次重复使用,限制了其应用。
技术实现要素:
本发明的目的是针对现有技术的不足,提供一种简单、快捷、易操作的壳聚糖基铅离子印迹纳米纤维的制备方法。
本发明是通过以下技术方案实现的:
一种壳聚糖基铅离子印迹纳米纤维的制备方法,其包括如下步骤:
s1、将聚苯乙烯加入n,n-二甲基甲酰胺和n-甲基吡咯烷酮的混合溶剂中,溶解后,在-50~-10℃下进行热致相分离;
s2、将步骤s1得到的产物浸泡于甲醇中,进行非溶剂致相分离后,冷冻干燥得到聚苯乙烯纳米纤维;
s3、将所述聚苯乙烯纳米纤维二苯酮的乙醇溶液中,活化后备用;
s4、将壳聚糖溶解于丙烯酸中,加入硝酸铅,混合均匀后,加入活化后的聚苯乙烯纳米纤维,用紫外光进行辐照聚合后,用戊二醛进行交联,得到壳聚糖基铅离子印迹纳米纤维前驱体;
s5、将所述壳聚糖基铅离子印迹纳米纤维前驱体用盐酸进行洗涤,除去铅离子,然后用蒸馏水洗涤除去盐酸,真空干燥,得到所述壳聚糖基铅离子印迹纳米纤维。
作为优选方案,步骤s1中所述的聚苯乙烯、n,n-二甲基甲酰胺和n-甲基吡咯烷酮的质量比为(4~8):(60~80):(15~30)。
作为优选方案,所述二苯酮的乙醇溶液中,二苯酮的质量分数为4~6%。
作为优选方案,步骤s4中所述的壳聚糖、丙烯酸和硝酸铅的质量比为(1~2):
(0.5~1):(0.2~0.3)。
作为优选方案,步骤s4中所述的紫外光的功率为500w,辐照聚合的时间为5min。
作为优选方案,所述戊二醛水溶液的质量分数为3~5%。
本发明的机理在于:
通过热致相分离和非溶剂致相分离结合制备聚苯乙烯纳米纤维。以聚苯乙烯纳米纤维为载体,将吸附铅离子的丙烯酸和壳聚糖螯合物,通过光引发接枝聚合到纳米纤维上。将接枝物通过戊二醛交联、盐酸洗涤得到壳聚糖基铅离子印迹纳米纤维。利用聚苯乙烯纳米纤维的大比表面积和高孔隙率,利用壳聚糖上氨基和丙烯酸上羟基的亲水性,大大提高了印迹纳米纤维的吸附速率和吸附容量。
与现有技术相比,本发明具有如下的有益效果:
1、壳聚糖基铅离子印迹纳米纤维作为铅离子的选择吸附剂,以纤维形式存在,有利于回收二次重复使用。
2、利用聚苯乙烯纳米纤维具有孔隙率高、比表面积大等优点,将具有重金属螯合功能的壳聚糖和丙烯酸接枝到纳米纤维上,利用离子印迹技术在纤维上引入铅离子识别位点,在保留纳米纤维优点的基础上,赋予其对铅离子高选择性分离的能力。
附图说明
通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
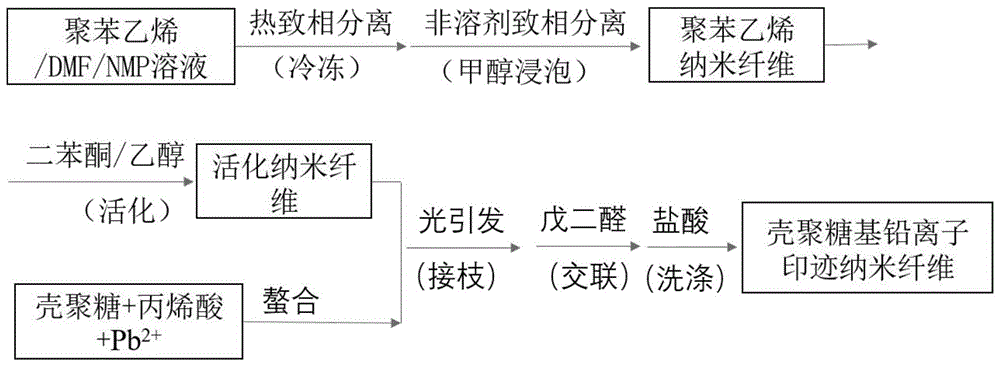
图1本发明的壳聚糖基铅离子印迹纳米纤维制备流程图;
图2本发明实施例1制备的壳聚糖基铅离子印迹纳米纤维扫描电镜图;
图3本发明实施例1制备的壳聚糖基铅离子印迹纳米纤维吸附容量与时间关系曲线;
图4本发明实施例1制备的壳聚糖基铅离子印迹纳米纤维吸附容量与温度关系曲线;
图5本发明对比例1制备的壳聚糖基铅离子印迹纳米纤维扫描电镜图。
具体实施方式
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进。这些都属于本发明的保护范围。
名词解释:
非溶剂:
聚合物的不良溶剂,且能和聚合物溶液中的溶剂互溶。
非溶剂致相分离:
将聚合物溶解在良溶剂中,后将聚合物溶液浸泡在非溶剂中,通过非溶剂诱导溶液,由于非溶剂与溶剂的相互交换发生相分离的过程。
实施例1
1)将0.5g聚苯乙烯加入由5gn,n-二甲基甲酰胺(dmp)和2.5gn-甲基吡咯烷酮(nmp)的混合溶剂中,常温下磁力搅拌6h,使其充分溶解得到溶液。将溶液倒入培养皿中,放入-40℃低温冰箱中冷冻120min(热致相分离),冷冻结束后快速拿出培养皿,放入500ml的甲醇溶液中(非溶剂致相分离),除去n,n-二甲基甲酰胺和n-甲基吡咯烷酮溶剂,每6h换甲醇一次,换4次,最后将样品冷冻干燥得到聚苯乙烯纳米纤维。
2)将聚苯乙烯纳米纤维浸泡在由4g二苯酮和96g乙醇组成的溶液中,10min后取出,真空干燥,得到活化的聚苯乙烯纳米纤维,备用。
3)将1g壳聚糖加入100ml蒸馏水中,磁力搅拌2h,后加入0.5g丙烯酸,磁力搅拌5h,形成均相溶液。将0.2g硝酸铅溶解在3ml蒸馏水中。将壳聚糖、丙烯酸溶液和硝酸铅溶液混合,常温下磁力搅拌6h。
4)将活化的聚苯乙烯纳米纤维浸泡在步骤3)制备的溶液中,500w紫外光照射10min取出。接着浸泡在50ml质量浓度为4%的戊二醛水溶液中,40℃反应2h。浸泡结束后,取出纤维用1mol/l盐酸反复洗涤,除去模板pb2+,最后用大量蒸馏水洗涤以除去残留的盐酸,真空干燥至恒重,得到壳聚糖基铅离子印迹纳米纤维,制备流程图如图1所示。
实施例1制备得到的壳聚糖基铅离子印迹纳米纤维的直径为216±83nm,如图2所示。孔隙率和比表面积分别为92.8%和6.78m2/g。
图3为壳聚糖基铅离子印迹纳米纤维对铅离子的吸附容量与溶液ph值的关系曲线图。在ph为1-2时,吸附容量约为30mg/g,随着ph值增大,吸附容量逐渐增加,当ph=5时,吸附容量达到最大值为158.13mg/g。ph值进一步增大,吸附容量逐渐降低。主要因为在较低ph条件下,印迹纳米纤维上的氨基和羧基质子化,使得吸附剂与铅离子之间产生静电排斥,吸附容量降低。随着ph值增大,质子化变弱,螯合作用逐渐变强,吸附容量逐渐增大。而ph值进一步升高时,铅离子易于与氢氧根发生沉淀。因此该吸附剂比较合适的吸附条件为ph=5。图4为壳聚糖基铅离子印迹纳米纤维和非印迹纳米纤维对铅离子吸附的动力学曲线。印迹纳米纤维和非印迹纳米纤维开始吸附阶段,随着吸附时间增加,吸附容量逐渐增大,30min后基本上达到吸附平衡。说明两种纳米纤维对铅离子的吸附速率很快,主要因为两者吸附剂都为纳米纤维结构,纤维与纤维之间含有大量的孔,且吸附剂上含有大量的亲水基团氨基和羧基,使溶液能快速在吸附剂上渗透,因此其在较短时间30min即可达到吸附平衡。非印迹纳米纤维的吸附动力学曲线虽然与印迹纳米纤维相似,但其平衡吸附量只有67.04mg/g,吸附容量大大降低。主要因为非印迹纳米纤维上不存在与铅离子具有尺寸大小、空间结构和结合位点分布相互匹配的孔穴,因此吸附容量大大降低。
实施例1制备的壳聚糖基铅离子印迹纳米纤维的最大吸附容量为158.13mg/g。印迹因子为印迹纳米纤维与非印迹纳米纤维最大吸附容量的比值。实施例1制备的壳聚糖基铅离子印迹纳米纤维的印迹因子为2.55。
将壳聚糖基铅离子印迹纳米纤维浸泡在pb2+和cu2+的混合溶液中,壳聚糖基铅离子印迹纳米纤维对pb2+/cu2+的选择性因子为3.01(选择性因子为印迹纤维对pb2+的最大吸附容量与cu2+最大吸附容量的比值)。说明该印迹纳米纤维对铅离子具有特定的选择性。
实施例2
1)将0.5g聚苯乙烯加入由9gn,n-二甲基甲酰胺(dmp)和2.5gn-甲基吡咯烷酮(nmp)的混合溶剂中,常温下磁力搅拌6h,使其充分溶解得到溶液。将溶液倒入培养皿中,放入-20℃低温冰箱中冷冻80min(热致相分离),冷冻结束后快速拿出培养皿,放入500ml的甲醇溶液中(非溶剂致相分离),除去n,n-二甲基甲酰胺和n-甲基吡咯烷酮溶剂,每6h换甲醇一次,换4次,最后将样品冷冻干燥得到聚苯乙烯纳米纤维。
2)将聚苯乙烯纳米纤维浸泡在由5g二苯酮和95g乙醇组成的混合溶液中,10min后取出,真空干燥,得到活化的聚苯乙烯纳米纤维,备用。
3)将2g壳聚糖加入100ml蒸馏水中,磁力搅拌2h,后加入1g丙烯酸,磁力搅拌5h,形成均相溶液。将0.25g硝酸铅溶解在3ml蒸馏水中。将壳聚糖、丙烯酸溶液和硝酸铅溶液混合,常温下磁力搅拌6h。
4)将活化的聚苯乙烯纳米纤维浸泡在步骤3)制备的溶液中,500w紫外光照射10min取出。接着浸泡在50ml质量浓度为5%的戊二醛水溶液中,40℃反应2h。浸泡结束后,取出纤维用1mol/l盐酸反复洗涤,除去模板pb2+,最后用大量蒸馏水洗涤以除去残留的盐酸,真空干燥至恒重,得到壳聚糖基铅离子印迹纳米纤维。
实施例2制备得到的壳聚糖基铅离子印迹纳米纤维的直径为198±77nm,孔隙率和比表面积分别为91.9%和6.67m2/g。实施例2制备的壳聚糖基铅离子印迹纳米纤维的最大吸附容量为156.28mg/g,印迹因子为2.59,对pb2+/cu2+的选择性因子为2.98。
实施例3
1)将0.5g聚苯乙烯加入由7gn,n-二甲基甲酰胺(dmp)和1.3gn-甲基吡咯烷酮(nmp)的混合溶剂中,常温下磁力搅拌6h,使其充分溶解得到溶液。将溶液倒入培养皿中,放入-30℃低温冰箱中冷冻90min(热致相分离),冷冻结束后快速拿出培养皿,放入500ml的甲醇溶液中(非溶剂致相分离),除去n,n-二甲基甲酰胺和n-甲基吡咯烷酮溶剂,每6h换甲醇一次,换4次,最后将样品冷冻干燥得到聚苯乙烯纳米纤维。
2)将聚苯乙烯纳米纤维浸泡在由6g二苯酮和96g乙醇组成的混合溶液中,10min后取出,真空干燥,得到活化的聚苯乙烯纳米纤维,备用。
3)将1.5g壳聚糖加入100ml蒸馏水中,磁力搅拌2h,后加入0.75g丙烯酸,磁力搅拌5h,形成均相溶液。将0.2g硝酸铅溶解在3ml蒸馏水中。将壳聚糖、丙烯酸溶液和硝酸铅溶液混合,常温下磁力搅拌6h。
4)将活化的聚苯乙烯纳米纤维浸泡在步骤3)制备的溶液中,500w紫外光照射10min取出。接着浸泡在50ml质量浓度为4%的戊二醛水溶液中,40℃反应2h。浸泡结束后,取出纤维用1mol/l盐酸反复洗涤,除去模板pb2+,最后用大量蒸馏水洗涤以除去残留的盐酸,真空干燥至恒重,得到壳聚糖基铅离子印迹纳米纤维。
实施例3制备得到的壳聚糖基铅离子印迹纳米纤维的直径为219±101nm,孔隙率和比表面积分别为92.8%和5.98m2/g。实施例3制备的壳聚糖基铅离子印迹纳米纤维的最大吸附容量为149.09mg/g,印迹因子为2.33,对pb2+/cu2+的选择性因子为2.81。
实施例4
1)将0.5g聚苯乙烯加入由5gn,n-二甲基甲酰胺(dmp)和2.2gn-甲基吡咯烷酮(nmp)的混合溶剂中,常温下磁力搅拌6h,使其充分溶解得到溶液。将溶液倒入培养皿中,放入-10℃低温冰箱中冷冻100min(热致相分离),冷冻结束后快速拿出培养皿,放入500ml的甲醇混合液中(非溶剂致相分离),除去n,n-二甲基甲酰胺和n-甲基吡咯烷酮溶剂,每6h换甲醇一次,换4次,最后将样品冷冻干燥得到聚苯乙烯纳米纤维。
2)将聚苯乙烯纳米纤维浸泡在由6g二苯酮和96g乙醇组成的混合溶液中,10min后取出,真空干燥,得到活化的聚苯乙烯纳米纤维,备用。
3)将2g壳聚糖加入100ml蒸馏水中,磁力搅拌2h,后加入1g丙烯酸,磁力搅拌5h,形成均相溶液。将0.3g硝酸铅溶解在3ml蒸馏水中。将壳聚糖、丙烯酸溶液和硝酸铅溶液混合,常温下磁力搅拌6h。
4)将活化的聚苯乙烯纳米纤维浸泡在步骤3)制备的溶液中,500w紫外光照射10min取出。接着浸泡在50ml质量浓度为5%的戊二醛水溶液中,40℃反应2h。浸泡结束后,取出纤维用1mol/l盐酸反复洗涤,除去模板pb2+,最后用大量蒸馏水洗涤以除去残留的盐酸,真空干燥至恒重,得到壳聚糖基铅离子印迹纳米纤维。
实施例4制备得到的壳聚糖基铅离子印迹纳米纤维的直径为210±78nm,孔隙率和比表面积分别为89.5%和6.34m2/g。实施例4制备的壳聚糖基铅离子印迹纳米纤维的最大吸附容量为153.20mg/g,印迹因子为2.53,对pb2+/cu2+的选择性因子为2.82。
对比例1
与实施例1不同之处在于:步骤1)中低温冰箱的淬火温度为15℃。最终得到铅离子印迹膜。膜的表面光滑,没有孔结构。印迹膜的孔隙率和比表面积分别为28.8%和0.41m2/g。印迹膜对铅离子的最大吸附容量为10.78mg/g,印迹因子为0.16,相对选择系数为2.21。无法得到纳米纤维结构主要因为冷冻温度高,溶液无法进行相分离,形成聚合物富集相和溶剂富集相,最终除去溶剂后只能得到光滑膜结构。吸附容量的降低主要因为制备的印迹膜为光滑膜结构,孔隙率和比表面积小,吸附活性点大大减少。
对比例2
与实施例1不同之处在于:没有制备聚苯乙烯纳米纤维,而是直接将步骤3)配制的壳聚糖、丙烯酸和硝酸铅溶液中加入0.2g过硫酸钾,在80℃反应2h,使丙烯酸发生聚合,聚合物结束后离心。将沉淀物浸泡在50ml质量浓度为5%的戊二醛水溶液中,40℃反应2h。浸泡结束后,取出沉淀物用1mol/l盐酸反复洗涤,除去模板pb2+,最后用大量蒸馏水洗涤以除去残留的盐酸,真空干燥至恒重,得到壳聚糖基铅离子印迹颗粒。壳聚糖基铅离子印迹颗粒的孔隙率为21.7%,比表面积0.57m2/g。
壳聚糖基铅离子印迹颗粒对铅离子的最大吸附容量为20.45mg/g。壳聚糖基铅离子印迹颗粒吸附容量大大降低,主要因为没有聚苯乙烯纳米纤维为载体,印迹颗粒的比表面积和孔隙率大大降低,是印迹颗粒上的氨基和羧基活性点降低,因此吸附容量降低。
对比例3
与实施例1不同之处在于:步骤3)中硝酸铅的添加量为0,最终得到非离子印迹纳米纤维。非离子印迹纳米纤维的直径为220±78nm,孔隙率和比表面积分别为91.2%和6.78m2/g。对比例3制备的非离子印迹纳米纤维的最大吸附容量为67.04mg/g,如图2所示。印迹因子为1,相对选择系数为0.89。虽然非印迹纳米纤维的孔隙率和比表面积没有降低,但是其对铅离子的吸附容量却大大降低。主要因为非印迹纳米纤维上不存在与铅离子具有尺寸大小、空间结构和结合位点分布相互匹配的孔穴,因此吸附容量大大降低。
对比例4
与实施例1不同之处在于:步骤1)中冷冻结束后快速拿出培养皿,放入500ml的蒸馏水中,除去n,n-二甲基甲酰胺和n-甲基吡咯烷酮溶剂,每6h换蒸馏水一次,换4次,最后将样品冷冻干燥。即以上过程溶液只经过热致相分离,没有非溶剂致相分离。最终得到铅离子印迹纤维。纤维的直径为610±141nm,且部分纤维发生了一定程度的粘连,如图5所示,印迹膜的孔隙率和比表面积分别为55.3%和1.38m2/g。印迹膜对铅离子的最大吸附容量为31.81mg/g。相比较于实施例1,纤维的直径大约为原来的2.7倍,孔隙率和比表面积也大大减小。主要因为冷冻热致相分离结束后,基本上形成纤维形貌,然而浸泡在蒸馏水中,形成的纤维发生了一定程度的粘连,使纤维与纤维之间的孔消失,因此比表面积和孔隙率减小,吸附容量大大降低。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变形或修改,这并不影响本发明的实质内容。
- 还没有人留言评论。精彩留言会获得点赞!