一种小干扰核酸药物组合物及其用途的制作方法
本发明涉及生物医药
技术领域:
,具体地,涉及一种小干扰核酸药物组合物及其用途。
背景技术:
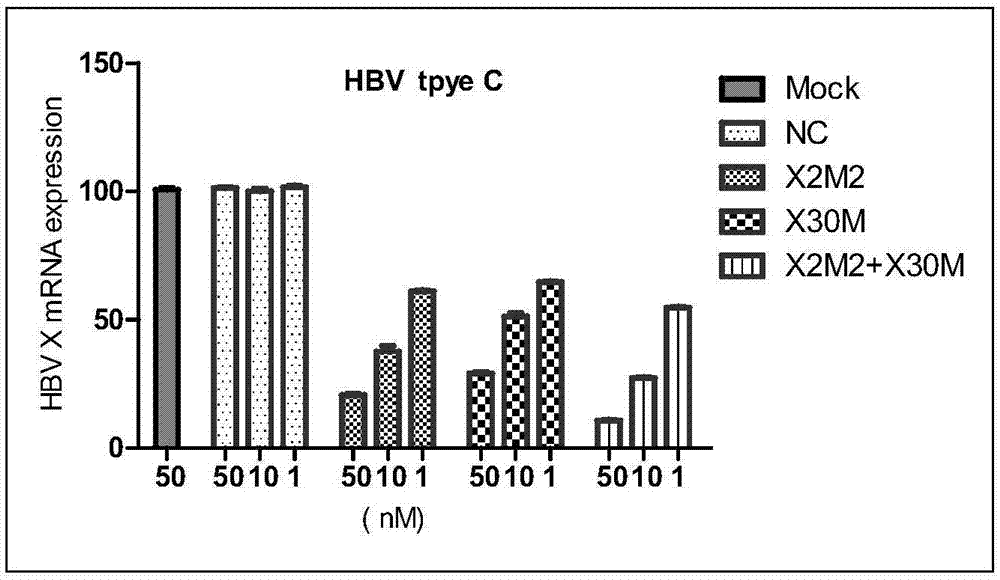
:乙型病毒性肝炎(又称乙型肝炎或乙肝)是严重威胁全球、特别是中国的一类传染病。HBV根据基因型可分为A、B、C、D、E、F、G、H这8种类型。这些基因型在序列上至少存在8%的差异,且具有不同的地域分布特点,其中,A型为全世界分布;B型、C型主要分布在亚洲;D型主要分布在美洲、南欧、澳大利亚和中东;E型主要分布在非洲;F型分布于美洲土著人和波利尼西亚;G型分布在美洲;H型在美国发现。目前全球公认的干扰素和核苷类似物这两大类乙肝防治药物仍存在多种弊端,如核苷类药物存在耐药性和停药后复发问题、干扰素易产生不良反应等。此外,最近的研究表明干扰素类药物对不同的HBV基因型表现出明显差别化的治疗反应率,如对B型的治疗反应率要明显高于C型(41%vs.15%)。核苷类药物也存在类似的发现。换言之,这两大类目前流行的乙肝防治药物仅针对特定基因型的患者才能发挥作用,具有很大局限性。所以,如果一种抗乙肝药物能涵盖不同的基因型,便可以提高对各基因型患者的治疗反应率,这将会给乙肝患者带来福音。小干扰RNA(smallinterferingRNA,siRNA)通过阻碍特定基因的翻译或转录来抑制基因表达。从理论上讲,siRNA可以通过序列特异性的方式抑制或阻断任何感兴趣的目的基因的表达,从而达到治疗疾病的目的。另一方面,对于乙型病毒性肝炎,传统药物只对HBVDNA有降低作用,对表面抗原的降低作用极其微弱,而siRNA药物则可以基于RNA干扰(RNAinterference,RNAi)机制从mRNA水平进行阻断,从而同时降低HBVDNA和HBsAg的水平。然而,由于siRNA通过序列特异性的方式发挥作用,鲜见有一种siRNA药物能够覆盖多种基因型。相应地,目前临床上迫切需要这样一种能同时针对HBV多种基因型并对HBsAg也具有抑制作用的药物。技术实现要素:本发明的目的是提供一种针对HBV基因的高效siRNA组合物,更优的,提供一种针对HBV多种基因型的药物组合物,该药物组合物有效预防和/或治疗病毒性乙型肝炎。为了实现上述目的,本发明第一方面提供了一种小干扰核酸药物组合物,该药物组合物含有siRNAX2M2和siRNAX30M,其中,siRNAX2M2的正义链含有如SEQIDNO:5所示的核苷酸序列,反义链含有如SEQIDNO:6所示的核苷酸序列;siRNAX30M的正义链含有如SEQIDNO:7所示的核苷酸序列,反义链含有如SEQIDNO:8所示的核苷酸序列;siRNAX2M2:正义链:5’-CCUUGAGGCAUACUUCAAA-3’(SEQIDNO:5),反义链:5’-UUUGAAGUAUGCCUCAAGG-3’(SEQIDNO:6),siRNAX30M:正义链:5’-CUAGGAGGCUGUAGGCAUA-3’(SEQIDNO:7),反义链:5’-UAUGCCUACAGCCUCCUAG-3’(SEQIDNO:8),其中,大写字母C、G、U和A分别表示核苷酸的碱基组成。第二方面,本发明提供了一种试剂盒,所述试剂盒含有如第一方面所述的药物组合物。第三方面,本发明提供了如第一方面所述的药物组合物在制备用于预防和/或治疗乙肝的药物中的用途。第四方面,本发明提供了一种治疗乙肝的方法,所述方法包括将如第一方面所述的药物组合物给予有需要的患者。第五方面,本发明提供了一种抑制感染慢性HBV的肝炎细胞中HBV基因表达的方法,该方法包括将如第一方面所述的药物组合物导入所述感染慢性HBV的肝炎细胞。有益效果相比传统药物(干扰素、核苷类药物)在HBV不同基因型覆盖方面存在局限的劣势,本发明的小干扰核酸药物组合物能够同时针对多种HBV基因型(包括例如B、C、D型)有效发挥作用(在细胞水平,50nM的药物组合物(siRNAX2M2和X30M组合物,摩尔比1:1)对上述3种HBV基因型mRNA的抑制率均达到88%以上);并且,本发明的小干扰核酸药物组合物在乙肝模型小鼠中表现出优异的HBVmRNA抑制效率(单次给药,14天后mRNA的抑制率仍维持在92%以上),同时能够有效地降低表面抗原表达(单次给药,14天后HBsAg的抑制率维持在95%),显示出对慢性乙型肝炎良好的治疗作用。更加出乎意料的是,相比各单独的siRNAX2M2和siRNAX30M,联合使用siRNAX2M2和siRNAX30M在细胞水平和动物水平都显示出了协同效果。具体表现为,在细胞水平,50nM的转染浓度下,单独使用一种siRNA对3种HBV基因型mRNA的抑制率在70%-80%,而联合使用X2M2和X30M组合物(摩尔比1:1)对HBV不同基因型的mRNA抑制率高达88%以上。联合使用不同比例的X2M2和X30M组合物相比单独的siRNA对C型、D型HBVmRNA的抑制率相当或更好。在动物水平,联合使用X2M2和X30M组合物(摩尔比1:1)相对于单独siRNA给药,对mRNA和HBsAg的抑制效果,无论从抑制率而言,还是从抑制时长而言,都获得了显著优势,即联合使用X2M2和X30M组合物对mRNA和HBsAg的抑制率能稳定维持在90%以上,且持续作用时间长达14天以上。而单独使用其中一种siRNA对mRNA和HBsAg的抑制率在给药后3天略低于组合物的效果,但在单次给药后14天,单独使用一种siRNA的抑制作用明显低于组合物的效果。由此可见,本发明提供了一种更具临床应用潜能的小干扰核酸药物组合物。本发明的其它特征和优点将在随后的具体实施方式中详细说明。附图说明图1示出了表1中所列的测试siRNA与对照siRNA在分别用携带B、C和D三种HBV基因型的克隆质粒转染的HepG2细胞中对HBVmRNA的抑制作用。图2示出了表1中所列的测试siRNA与对照siRNA在单次给药后3天和14天对M-TgHBV转基因小鼠肝组织中HBVmRNA的抑制作用。图3示出了表1中所列的测试siRNA与对照siRNA在单次给药后3天和14天对M-TgHBV转基因小鼠血清中HBsAg的抑制作用。具体实施方式以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。一.本发明的小干扰核酸药物组合物本发明提供了一种小干扰核酸药物组合物,其中,所述药物组合物含有siRNAX2M2和siRNAX30M,其中,siRNAX2M2的正义链含有如SEQIDNO:5所示的核苷酸序列,反义链含有如SEQIDNO:6所示的核苷酸序列;siRNAX30M的正义链含有如SEQIDNO:7所示的核苷酸序列,反义链含有如SEQIDNO:8所示的核苷酸序列;siRNAX2M2:正义链:5’-CCUUGAGGCAUACUUCAAA-3’(SEQIDNO:5),反义链:5’-UUUGAAGUAUGCCUCAAGG-3’(SEQIDNO:6),siRNAX30M:正义链:5’-CUAGGAGGCUGUAGGCAUA-3’(SEQIDNO:7),反义链:5’-UAUGCCUACAGCCUCCUAG-3’(SEQIDNO:8),其中,大写字母C、G、U和A分别表示核苷酸的碱基组成。在本发明的小干扰核酸药物组合物中,以任意不同比例存在的siRNAX2M2和siRNAX30M组合物均能够达到本发明的目的。优选地,所述siRNAX2M2与所述siRNAX30M的摩尔比为1:100~100:1。为了体现更好的治疗效果和协同作用,按照本发明一个具体实施方式,siRNAX2M2与siRNAX30M的摩尔比可以为1:10-10:1、1:5-5:1或1:2-2:1。siRNAX2M2正义链和反义链可以是如SEQIDNO:5和如SEQIDNO:6所示的裸核苷酸序列;siRNAX30M正义链和反义链可以是如SEQIDNO:7和如SEQIDNO:8所示的裸核苷酸序列。优选情况下,为了增强siRNA双链的稳定性,siRNAX2M2的正义链和反义链以及siRNAX30M的正义链和反义链中至少一条单链的3’末端还连接有1至3个核苷酸,从而在所述正义链和所述反义链互补配对后形成至少一个由1至3个核苷酸构成的3’突出端,或称作悬垂末端。优选地,siRNAX2M2的正义链和反义链以及siRNAX30M的正义链和反义链中的每一条单链的3’末端均连接有3’突出端。更优选地,所述3’突出端为连续的2个脱氧胸腺嘧啶核苷酸(即dTdT)或连续的2个尿嘧啶核苷酸(即UU)。为了进一步提高siRNAX2M2和/或siRNAX30M的稳定性,所述siRNAX2M2和/或siRNAX30M的磷酸-糖骨架中的磷酸酯基中的至少一部分为具有修饰基团的磷酸酯基,所述siRNAX2M2和/或siRNAX30M的磷酸-糖骨架中的核糖基中的至少一部分为具有修饰基团的核糖基。具有修饰基团的磷酸酯基和核糖基可以是具有上述作用的各种具有修饰基团的磷酸酯基和核糖基。优选地,具有修饰基团的核糖基为2’-羟基被甲氧基取代而成的2’-甲氧基核糖基或者为2’-羟基被氟取代而成的2’-氟代核糖基,具有修饰基团的磷酸酯基为磷酸酯基中的磷酸二酯键中的一个氧原子被硫原子取代而成的硫代磷酸酯基;所述硫代磷酸酯基结构如式VI所示:按照本发明一个具体实施方式,本发明提供的小干扰核酸药物组合物中,siRNAX2M2的正义链为如SEQIDNO:1所示的核苷酸序列,反义链为如SEQIDNO:2所示的核苷酸序列;siRNAX30M的正义链为如SEQIDNO:3所示的核苷酸序列,反义链为如SEQIDNO:4所示的核苷酸序列:siRNAX2M2:正义链5’-CmCmUmUmGAGGCmAUmACmUmUmCmAAA-dT-s-dT-3’(SEQIDNO:1);反义链5’-UfUmUfGAAGUfAUGCCUfCAAGG-dT-s-dT-3’(SEQIDNO:2),siRNAX30M:正义链5’-CmUAmGGAGGCUmGmUAmGGCmAUmAdT-s-dT-3’(SEQIDNO:3),反义链5’-UAmUGCCUfACAGCCUCCUfAGdT-s-dT-3’(SEQIDNO:4),其中,小写字母m表示该字母m左侧的一个核苷酸的核糖基为2’-羟基被甲氧基取代而成的2’-甲氧基核糖基;小写字母f表示该字母f左侧的一个核苷酸的核糖基为2’-羟基被氟取代而成的2’-氟代核糖基;小写字母d表示该字母d右侧的一个核苷酸为脱氧核糖核苷酸,大写字母T表示核苷酸的碱基组成;小写字母s表示3’突出端dTdT之间的磷酸酯基为硫代磷酸酯基。例如,在siRNAX2M2中,由正义链5’端第1个大写字母C及其右侧的m组成的“Cm”表示所含碱基为胞嘧啶、核糖基的2’-羟基被甲氧基取代的核苷酸;由反义链5’端第1个大写字母U及其右侧的f组成的“Uf”表示所含碱基为尿嘧啶、核糖基的2’-羟基被氟取代的核苷酸。本领域技术人员清楚知晓的是,通过本领域常规的siRNA制备方法(例如固相合成和液相合成)可以得到本发明所述的siRNAX2M2和/或siRNAX30M,其中,固相合成已经有商业化订制服务。本领域技术人员也清楚知晓,通过使用具有相应修饰的核苷酸单体可以将修饰的核苷酸基团引入本公开所述的siRNAX2M2和/或siRNAX30M中。制备具有相应修饰的核苷酸单体的方法是本领域技术人员所熟知的,市场上也有商业化的单体供应。在本发明的小干扰核酸药物组合物中,除含有siRNAX2M2和siRNAX30M两种siRNA作为活性成分外,还任选地含有药学上可接受的载体。所述药学上可接受的载体可以是siRNA给药领域常规使用的载体,例如但不限于磁性纳米粒(magneticnanoparticles,如Fe3O4、Fe2O3)、碳纳米管(carbonnanotubes)、介孔硅(mesoporoussilicon)、磷酸钙纳米粒(calciumphosphatenanoparticles)、聚乙烯亚胺(polyethylenimine,PEI)、聚酰胺胺型树形高分子(polyamidoamine(PAMAM)dendrimer)、聚L-赖氨酸(poly(L-lysine),PLL)、壳聚糖(chitosan)、1,2-二油酰基-3-三甲铵丙烷(1,2-dioleoyl-3-trimethylammonium-propane,DOTAP)、聚D型或L型乳酸/羟基乙酸共聚物(poly(D&L-lactic/glycolicacid)copolymer,PLGA)、聚(2-氨乙基乙撑磷酸酯)(poly(2-aminoethylethylenephosphate),PPEEA)和聚(甲基丙烯酸-N,N-二甲氨基乙酯)(poly(2-dimethylaminoethylmethacrylate),PDMAEMA)以及它们的衍生物中的一种或多种。在本发明的小干扰核酸药物组合物中,对药学上可接受的载体的含量没有特别要求,一般地,本发明的小干扰核酸药物组合物中所含siRNAX2M2和siRNAX30M的总重量与药学上可接受的载体的重量比可为1:(1-500),优选为1:(1-50)。在本发明的小干扰核酸药物组合物中,还可以包含药学上可接受的其它辅料,该辅料可以为本领域常规采用的各种制剂或化合物的一种或多种。例如,所述药学上可接受的其它辅料可以包括pH值缓冲液、保护剂和渗透压调节剂中的至少一种。所述pH值缓冲液可以为pH值7.5-8.5的三羟甲基胺基甲烷盐酸盐缓冲液和/或pH值5.5-8.5的磷酸盐缓冲液,优选为pH值5.5-8.5的磷酸盐缓冲液。所述保护剂可以为肌醇、山梨醇、蔗糖、海藻糖、甘露糖、麦芽糖、乳糖和葡糖糖中的至少一种。以所述小干扰核酸药物组合物的总重量为基准,所述保护剂的含量可以为0.01-30重量%。所述渗透压调节剂可以为氯化钠和/或氯化钾。所述渗透压调节剂的含量使所述药物组合物的渗透压为200-700毫渗摩尔/千克。根据所需渗透压,本领域技术人员可以容易地确定所述渗透压调节剂的含量。在本发明的小干扰核酸药物组合物的一些实施方式中,所述小干扰核酸药物组合物可以为液体制剂,例如注射液;也可以为冻干粉针剂,实施给药时与液体辅料混合,配制成液体制剂。所述液体制剂可以但不限于用于皮下、肌肉或静脉注射给药,也可以但不限于通过喷雾给药到肺脏、或通过喷雾经肺脏给药到其它脏器组织(如肝脏)。优选地,所述小干扰核酸药物组合物用于静脉注射给药。在本发明的小干扰核酸药物组合物的一个优选实施方式中,所述小干扰核酸药物组合物可以为脂质体制剂的形式。在本发明的小干扰核酸药物组合物的一个更优选的实施方式中,所述脂质体制剂中使用的药学上可接受的载体包含含胺化合物、辅助脂质和/或聚乙二醇化脂质。其中,所述含胺化合物、辅助脂质和聚乙二醇化脂质可分别选自于CN201180060664.1(通过引用将其整体并入本文)中所描述的含胺的转染化合物或其药学上可接受的盐或衍生物、辅助脂质和聚乙二醇化脂质中的一种或多种。具体而言,所述含胺化合物可为CN201180060664.1中描述的如式I所示的化合物和/或其药学上可接受的盐:其中:X1和X2各自独立地是O、S、N-A或C-A,其中A是氢或C1-C20烃链;Y和Z各自独立地是C=O、C=S、S=O、CH-OH或SO2;R1、R2、R3、R4、R5、R6和R7各自独立地是氢,环状或无环的、被取代的或未被取代的、支链或直链脂族基团,环状或无环的、被取代的或未被取代的、支链或直链杂脂族基团,被取代的或未被取代的、支链或直链酰基,被取代的或未被取代的、支链或直链芳基,被取代的或未被取代的、支链或直链杂芳基;x是1-10的整数;n是1-3的整数,m是0-20的整数,p是0或1,其中,如果m=p=0,那么R2是氢,并且,如果n或m中的至少一个是2,那么R3和在式I中的氮形成如式II或式III所示的结构:其中,g、e和f各自独立地是1-6的整数,“HCC”代表烃链,且每个*表示在式I中的氮原子。在某些实施方式中,R3是多胺。在其它实施方式中,R3是缩酮。在某些实施方式中,在式I中的R1和R2中的每一个独立地是任意的被取代的或未被取代的、支链或直链烷基或烯基,所述烷基或烯基具有3至约20个碳原子,诸如8至约18个碳原子,和0至4个双键,诸如0至2个双键。在某些实施方式中,如果n和m中的每一个独立地具有1或3的值,R3是下述部分中的任一个:其中每个“HCC”代表烃链,且每个*显示R3与在式I中的氮原子的可能连接点,其中在任意*位置上的每个H可以被替换以实现与在式I中的氮原子的连接。其中,式I所示化合物可以根据CN201180060664.1中的描述制备。优选地,所述含胺化合物可为CN201180060664.1中描述的含胺化合物72(这里称为式IV)或含胺化合物87(这里称为式V),如下所示:优选地,所述辅助脂质可以是胆固醇、胆固醇的类似物和/或胆固醇的衍生物。优选地,所述聚乙二醇化脂质可以是1,2-二棕榈酰基-sn-甘油-3-磷脂酰乙醇胺-N-[甲氧基(聚乙二醇)-2000],即1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000]。在本发明的小干扰核酸药物组合物的一个更优选的实施方式中,所使用的药学上可接受的载体同时包含上文所描述的含胺化合物、辅助脂质、聚乙二醇化脂质这三种组分,这三种组分形成脂质混合物。在该优选的实施方式中,所述药物组合物中含胺化合物、辅助脂质、聚乙二醇化脂质三者之间的摩尔比为(19.7-80):(19.7-80):(0.3-50)。更优选地,所述药物组合物中含胺化合物、辅助脂质、聚乙二醇化脂质三者之间的摩尔比为(50-70):(20-40):(3-20)。由siRNAX2M2及siRNAX30M与上述脂质混合物形成的脂质体颗粒具有约30nm至约200nm的平均直径,通常为约40nm至约135nm,更通常地,该脂质体颗粒的平均直径是约50nm至约120nm、约50nm至约100nm、约60nm至约90nm或约70nm至约90nm,例如,该脂质体颗粒的平均直径是约30、40、50、60、70、75、80、85、90、100、110、120、130、140、150或160nm。在脂质体制剂形式的小干扰核酸药物组合物中,siRNAX2M2及siRNAX30M的总重量与全部脂质(例如含胺化合物、辅助脂质和/或聚乙二醇化脂质)的重量比(重量/重量比)在从约1:1至约1:50、从约1:1至约1:30、从约1:3至约1:20、从约1:4至约1:18、从约1:5至约1:17、从约1:5至约1:15、从约1:5至约1:12、从约1:6至约1:12或从约1:6至约1:10的范围内,例如,siRNAX2M2及siRNAX30M的总重量与全部脂质的重量比为约1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14、1:15、1:16、1:17或1:18。本发明提供的药物组合物在销售时各组分可以独立存在,在使用时可以液体制剂的形式存在。本发明提供的含有上述药学上可接受的载体形成的药物组合物可以按照已知的各种方法制备。按照本发明一个实施方式,本发明提供的药物组合物与上述脂质混合物形成的药物组合物可以根据CN201180060664.1中的描述方法制备;更优选地,可以按照如下方法制备:将含胺化合物、辅助脂质和聚乙二醇化脂质按照上述摩尔比悬浮于醇中并混匀得到脂质溶液;醇的用量使得到的脂质溶液的总质量浓度为2-25mg/mL,优选为8-18mg/mL。所述醇选自药学上可接受的醇,诸如在室温附近为液体的醇,例如,乙醇、丙二醇、苯甲醇、甘油、聚乙二醇200、聚乙二醇300、聚乙二醇400中的一种或多种,优选为乙醇。将siRNAX2M2和siRNAX30M溶解于缓冲盐溶液中,得到siRNA水溶液。缓冲盐溶液的浓度为0.05-0.5M,优选为0.1-0.2M,调节缓冲盐溶液的pH至4.0-5.5,优选为5.0-5.2,缓冲盐溶液的用量使siRNA的浓度不超过0.6mg/mL,优选为0.2-0.4mg/mL。所述缓冲盐选自可溶性醋酸盐、可溶性柠檬酸盐中的一种或多种,优选为醋酸钠和/或醋酸钾。将脂质溶液和siRNA水溶液混合,将混合后得到的产物在40-60℃孵育至少2分钟,优选5-30分钟,得到孵育后的脂质体制剂。脂质溶液和siRNA水溶液的体积比为1:(2-5),优选1:3。将孵育后的脂质体制剂浓缩或稀释,去除杂质,除菌,得到本发明提供的药物组合物,其理化参数为pH值为6.5-8,包封率不低于80%,粒径为40-200nm,多分散指数不高于0.30,渗透压为250-400mOsm/kg;优选地,pH值为7.2-7.6,包封率不低于90%,粒径为60-100nm,多分散指数不高于0.20,渗透压为300-400mOsm/kg。其中,浓缩或稀释可以在去除杂质之前、之后或同时进行。去除杂质的方法可以采用现有各种方法,优选使用切相流系统,中空纤维柱,在100KDa条件下超滤,超滤交换溶液为pH7.4的磷酸盐缓冲液(PBS)。除菌的方法可以采用现有各种方法,优选在0.22μm滤器上过滤除菌。二.本发明的试剂盒本发明提供的试剂盒含有如上所述本发明提供的药物组合物。其中,siRNAX2M2和siRNAX30M可以单独存在,也可以以两种的混合物的形式存在。在含有药学上可接受的载体和/或辅料的情况下,载体和/或辅料可以单独存在,以其中两种或多种的混合物的形式存在,或者以最终药物组合物的形式存在。当药学上可接受的载体单独存在且所述载体为上述脂质混合物时,含胺化合物、辅助脂质和聚乙二醇化脂质可以各自独立存在,也可以以其中两种或三种的混合物的形式存在。在一个实施方式中,可使一个容器用于提供siRNA、另外一个或多个容器用于提供含胺化合物、辅助脂质和聚乙二醇化脂质,任选地另外一个或多个容器提供辅料。除了siRNA和药学上可接受的载体和/或辅料以外,所述试剂盒中还可包含实现本发明提供的药物组合物的一种或多种特定应用所必需或有益的组分,如(1)一种或多种用于实现所希望的细胞转染的组分,(2)一种或多种用于实现特定疾病或身体障碍的诊断、治疗或预防的组分,如一种或多种额外的治疗化合物或组合物、一种或多种诊断试剂,(3)一种或多种缓冲剂,(4)阳性或阴性对照样品,(5)赋形剂、稳定剂或防腐剂等。一般来说,所述组分存在于与siRNA和药学上可接受的载体和/或辅料的容器均不同的容器中。此外,所述试剂盒还可包含用于将siRNAX2M2和/或siRNAX30M与药学上可接受的载体和/或辅料或其它成分进行混合的说明书。在本发明提供的试剂盒中,药物组合物可以任何形式提供,例如液体形式、干燥形式或冻干形式。优选情况下,所述药物组合物基本上纯净和/或无菌。可任选地在本发明的试剂盒中提供无菌水、生理盐水、PBS中的一种或多种。三.本发明的小干扰核酸药物组合物的用途本发明提供了如上所述的药物组合物在制备用于预防和/或治疗乙肝的药物中的用途。本发明还提供了一种预防和/或治疗乙肝的方法,包括将所述小干扰核酸药物组合物给予有需要的患者,通过RNAi机制达到治疗乙肝的目的。本文所使用的术语“给药/给予”是指通过使得至少部分地将siRNA或小干扰核酸药物组合物定位于期望的位点以产生期望效果的方法或途径,将siRNA或小干扰核酸药物组合物放置入受试者体内。适于本发明方法的给药途径包括局部给药和全身给药。一般而言,局部给药导致与受试者整个身体相比将更多siRNA或小干扰核酸药物组合物递送至特定位点;而全身给药导致将所述siRNA或小干扰核酸药物组合物递送至受试者的基本整个身体。考虑到本发明旨在提供用于预防和/或治疗乙肝的手段,优选能够将药物特异性递送至肝脏的给药方式。可通过本领域已知的任何合适途径向受试者进行给药,所述途径包括但不仅限于:口服或胃肠外途径,包括静脉内给药、肌肉内给药、皮下给药、经皮给药、气道给药(气雾剂)、肺部给药、鼻部给药、直肠给药和局部给药(包括口腔含化给药和舌下给药)。给药频率可以是每天、每周、每个月或每年1次或多次。本发明所述的小干扰核酸药物组合物的使用剂量可为本领域常规的剂量,所述剂量可以根据各种参数、尤其是受试者的年龄、体重和性别来确定。可在细胞培养或实验动物中通过标准药学程序测定毒性和疗效,例如测定LD50(使50%的群体致死的剂量)和ED50(在量反应中指能引起50%最大反应强度的剂量,在质反应中指引起50%实验对象出现阳性反应时的剂量)。毒性和疗效之间的剂量比为治疗指数,可以用LD50/ED50的比值来表示。优选显示出高治疗指数的siRNA或药物组合物。可基于由细胞培养分析和动物研究得到的数据得出人用剂量的范围。在给予本发明所述的小干扰核酸药物组合物时,例如,对于雄性或雌性、6-12周龄、体重18-25g的C57BL/6J或C3H/HeNCrlVr小鼠,以所述药物组合物中的siRNA的量计:对于siRNA与药学上可接受的载体形成的药物组合物,其siRNA用量可以为0.001-50mg/kg体重,优选为0.01-10mg/kg体重,更优选为0.05-5mg/kg体重,最优选为0.1-3mg/kg体重。在给予本发明的小干扰核酸药物组合物时,可参考上述用量。根据本发明的另一方面,通过将本发明的小干扰核酸药物组合物导入感染慢性HBV的肝炎细胞,还可以通过RNAi机制达到抑制感染慢性HBV的肝炎细胞中的HBV基因表达这一目的。在某些实施方式中,所述细胞为HepG2.2.15细胞,或表达有某种HBV基因型的HepG2细胞。采用本发明提供的方法抑制HBV基因在肝炎细胞中表达,无论使用提供的siRNA还是药物组合物,siRNA用量一般是这样的量:其足以减少靶基因的表达,并导致在靶细胞表面处100pM至1μM、或1nM至100nM、或5nM至50nM或至约10nM的细胞外浓度。达到该局部浓度所需的量将随各种因素而变化,所述因素包括递送方法、在递送部位和靶细胞或组织之间的细胞层的数目、递送是局部还是全身等。在递送部位处的浓度可以显著高于在靶细胞或组织的表面处的浓度。实施例通过以下实施例对本发明进行进一步详细的说明,但本发明不仅限于这些实施例。需要说明的是,在下述实施例中,虽然仅通过经过修饰的siRNAX2M2和siRNAX30M对本发明进行说明,但是,含有不经过修饰的siRNAX2M2和siRNAX30M的药物组合物也具有活性和一定的稳定性。除非特别说明,以下实施例所用到的试剂、培养基均为市售商品,所用到的核酸电泳、实时PCR等操作均按常规方案进行。例如,可按MolecularCloning(ColdSpringHarborLaboratoryPress(1989))所记载的进行。制备实施例1通过常规固相合成方法得到表1中所列的siRNA。用退火盐溶液溶解等摩尔的正义链和反义链混合物,随后常规退火以形成siRNA双链。其中,NC为与HBVmRNA无靶向作用的无关序列,作为阴性对照,下文也将其称为对照siRNA;其余siRNA(siRNAX2M2及siRNAX30M)则为特异性靶向HBV的siRNA,下文也将其称为测试siRNA。表1siRNA的序列注:大写字母C、G、U、A和T表示核苷酸的碱基组成;小写字母d表示该字母d右侧的一个核苷酸为脱氧核糖核苷酸;小写字母m表示该字母m左侧的一个核苷酸的核糖基为2’-羟基被甲氧基取代而成的2’-甲氧基核糖基;小写字母f表示该字母f左侧的一个核苷酸的核糖基为2’-羟基被氟取代而成的2’-氟代核糖基;小写字母s表示3’突出端dTdT之间的磷酸酯基为硫代磷酸酯基。制备实施例2将三种干粉脂质化合物(即,含胺化合物、辅助脂质、聚乙二醇化脂质)以59:29:12的摩尔比悬浮于乙醇中并混合,三种脂质化合物的总质量浓度约为8.85mg/ml。将表1所列的已合成的siRNA溶解于200mM醋酸钠(pH5.2)溶液中,使siRNA的浓度为0.2mg/ml(对于单独应用siRNAX2M2及siRNAX30M的情况,使siRNA的浓度为0.2mg/ml;对于组合应用siRNAX2M2及siRNAX30M的情况,将两种siRNA等比例混合,即,各siRNA的浓度分别为0.1mg/ml)。以1:3的体积比,非常迅速地混合所得到的脂质乙醇溶液和siRNA醋酸钠水溶液。在表2中描述了混合后得到的脂质体制剂的具体组成。表2脂质体制剂的组成将混合后得到的脂质体制剂(即,含胺化合物、胆固醇、聚乙二醇化脂质与siRNA的组合物)在约50℃孵育10分钟。孵育后,使用切相流系统,中空纤维柱100KDa超滤,超滤交换溶液为pH7.4的PBS。超滤的同时可以将制剂浓缩或稀释到期望的siRNA浓度。超滤后的制剂在0.22μm滤器上过滤除菌。将由含胺化合物式V、胆固醇、1,2-二棕榈酰基-sn-甘油-3-磷脂酰乙醇胺-N-[甲氧基(聚乙二醇)-2000]组成的脂质混合物称为RBP131。将由含胺化合物式IV、胆固醇、1,2-二棕榈酰基-sn-甘油-3-磷脂酰乙醇胺-N-[甲氧基(聚乙二醇)-2000]组成的脂质混合物称为RBP130。所得药物组合物RBP131/siRNA或RBP130/siRNA在使用前储存在4℃,并检测相关理化性质,RBP131/siRNA和RBP130/siRNA的理化参数相似,检测结果见表3。表3药物组合物的理化参数检测指征结果pH7.2-7.6包封率(%)≥90%siRNA浓度(mg/ml)0.10-0.15粒径(nm)60-100PDI≤0.2渗透压(mOsm/kg)300-400其中,包封率采用RiboGreen法检测,所用试剂(Quant-iTTMRNAReagentandKit)购于ThermoFisher(Invitrogen)公司,货号R11490。根据说明书操作步骤检测样品中siRNA的荧光强度,再按照文献(J.Heyeset.al,JournalofControlledRelease,107(2005):276–287)所述方法计算包封率:包封率=[(Triton处理组荧光强度-无Triton处理组荧光强度)/Triton处理组荧光强度]×100%其他理化参数采用本领域技术人员熟知的常规技术手段检测。实验实施例1本实验实施例采用携带B、C和D三种HBV基因型的克隆质粒在HepG2细胞上进行HBV的表达和复制,用来检测制备实施例1中得到的siRNA及其组合对HBVmRNA表达量的体外(invitro)抑制效率。其中,携带B、C和D三种HBV基因型的克隆质粒购自上海吉玛制药技术有限公司。使用携带B、C和D三种HBV基因型的克隆质粒分别转染HepG2细胞(购自ATCC)。转染前一天,取对数生长期细胞,以104个细胞/孔的密度接种于24孔板中,常规培养,当细胞密度达到80-90%时,更换为无双抗培养液。将携带B、C和D三种HBV基因型的克隆质粒以0.8μg/孔的浓度溶于50μlOpti-MEM(Gibco)中,轻轻混合。将用制备实施例1中得到的siRNA加入上述含不同基因型的克隆质粒溶液中,同时进行体外转染,每组3个平行(3个孔),至少重复3次实验。将1μLLipofectamineTM2000(Invitrogen公司)稀释于50μLOpti-MEM无血清培养基中,混匀后室温下孵育5分钟;混合稀释的上述克隆质粒与siRNA溶液和稀释的LipofectamineTM2000,轻轻混匀,室温静置20分钟,以便形成转染复合物。在接种有HepG2细胞的24孔板中按照每孔100μL加入上述最终混合溶液。siRNA终浓度分别为50nM、10nM、1nM。细胞于37℃培养4小时,再向每孔中加入1mL含有10%胎牛血清的DMEM完全培养基(Gibco),继续在37℃培养过夜。转染48小时后,采用实时荧光定量PCR(real-timefluorescentqPCR)测定以上收获的细胞中HBVmRNA的表达水平。具体地:使用RNeasyMiniKit(QIAGEN公司,货号Cat.74106)试剂盒,按其说明书进行总RNA提取;分别取1μg总RNA按照反转录试剂盒(Promega公司,货号A3500)的使用方法反转录得到cDNA。使用2×UltraSYBRMixture(withROX)(北京康为世纪生物科技有限公司,货号CW0956)试剂盒,以cDNA为模板按照说明书的步骤进行HBVmRNA的表达量检测。其中,用于扩增HBV和作为内参基因的GAPDH的PCR引物序列如下表4所示:表4检测引物的序列在该荧光定量PCR法中,siRNA抑制活性用HBV基因表达抑制率表示,按如下等式计算:HBV基因表达抑制率={1-(测试组HBV基因的拷贝数/测试组GAPDH基因的拷贝数)/(mock组HBV基因的拷贝数/mock组GAPDH基因的拷贝数)}×100%。其中,各测试组为分别经X2M2、X30M和X2M2+X30M处理的HepG2细胞,联合用药组(X2M2+X30M)中两个siRNA的摩尔比为1:1,两个siRNA的总用药量等同于单独的X2M2和X30M各自的用药量,例如,50nM联合用药组是指25nMX2M2+25nMX30M,其它浓度以此类推;mock组为经克隆载体转染但未经任何siRNA处理的HepG2细胞;NC组为经阴性对照siRNANC处理的HepG2细胞。结果如图1所示。图1显示了测试siRNA与对照siRNA在分别转染了携带HBV不同基因型的克隆质粒的HepG2细胞中对HBVmRNA表达抑制活性的检测结果。从图1的结果可以看出,单独的X2M2和X30M对HBVmRNA的抑制作用相当,而联合用药组X2M2+X30M在分别转染了携带不同HBV基因型的克隆质粒的HepG2细胞中对HBVmRNA的表达抑制作用均优于各单独用药组(即,对于HBV不同基因型均产生了协同效果),此结果为后续的联合用药提供了研究基础。实验实施例2本实验实施例采用携带C和D两种HBV基因型的克隆质粒分别在HepG2细胞上进行HBV的表达和复制,用来检测制备实施例1中得到的siRNA和不同比例的组合物(X2M2+X30M)对HBVmRNA表达量的体外(invitro)抑制效率。实验的具体方法参照实验实施例1,选择siRNA或组合物的终浓度为10nM进行测试,测试组为单独的X2M2和X30M,以及两者组合物(X2M2+X30M),其摩尔比分别为1:10、1:5、1:2、1:1、2:1、5:1、10:1,以上测试组合物中X2M2和X30M的含量如表5所示:表510nM组合物中各siRNA的含量组合物比例X2M2量(nM)X30M量(nM)1:100.919.091:51.678.331:23.336.671:1552:16.673.335:18.331.6710:19.090.91结果如表6所示,显示了测试siRNA与对照siRNA在分别转染了携带C或D两种不同基因型的克隆质粒的HepG2细胞中对HBVmRNA表达抑制活性的检测结果。表6测试siRNA或组合物对HBV基因表达抑制率其中,mock组为经克隆质粒转染但未经任何siRNA处理的HepG2细胞;NC组为经阴性对照siRNANC处理的HepG2细胞。从表6的结果可以看出,不论以何种比例进行组合,在分别转染C或D基因型的克隆质粒的HepG2细胞中,组合物对同一种基因型的HBV基因表达抑制率明显高于X2M2和X30M中的一种,与此同时高于或相当于另外一种,体现了两种siRNA良好的协同作用。实验实施例3本实验实施例用来检测制备实施例2中得到的RBP131/siRNA脂质体制剂在HBV转基因小鼠(命名为M-TgHBV,购自上海市公共卫生中心动物部,转基因小鼠的制备方法如RenJ.等,J.MedicalVirology.2006,78:551-560所述)中对HBVmRNA表达量和血清HBsAg的抑制效率。其中,所用siRNA同实验实施例1。将40只M-TgHBV小鼠按血清HBsAg含量随机分成10组(每组4只,均为雌性),分别为PBS对照组(3天和14天)、RBP131/siNC组(3天和14天)、RBP131/X2M21mg/kg组(3天和14天)、RBP131/X30M1mg/kg组(3天和14天)和RBP131/X2M2(0.5mg/kg)+X30M(0.5mg/kg)组(3天和14天)。以上每个组均设置有2个小组(即3天和14天处理组)。所有动物根据体重计算药量,尾静脉单次给药,给药体积为10ml/kg。给药后分别在第3天和14天将动物处死,处死前眼眶静脉丛取血约0.5ml,用于检测HBsAg的表达量;随后,对动物进行大体解剖,观察体内脏器是否有病变,对肉眼观察有病变的组织用10%福尔马林保存进一步进行病理观察,收集肝脏,用RNAlater(SigmaAldrich公司)保存。(1)肝组织mRNA检测。用组织匀浆仪匀浆肝组织,再用Trizol根据总RNA提取的标准操作步骤提取得到总RNA。采用实时荧光定量PCR检测肝组织中HBVmRNA的表达水平,具体地:使用ImProm-IITM反转录试剂盒(Promega公司)按其说明书将提取的总RNA逆转录为cDNA,接着用荧光定量PCR试剂盒(北京康为世纪生物科技有限公司)检测siRNA对肝组织中的HBVmRNA表达的抑制效率。在该荧光定量PCR法中,以β-肌动蛋白(β-actin)基因作为内参基因,使用针对HBV的引物和针对β-肌动蛋白的引物分别对HBV和β-肌动蛋白进行检测。检测引物的序列参见表7。表7检测引物的序列在该荧光定量PCR法中,siRNA抑制活性用HBV基因表达剩余量表示,按如下等式计算:HBV基因表达剩余量=(测试组HBV基因的拷贝数/测试组β-actin基因的拷贝数)/(对照组HBV基因的拷贝数/对照组β-actin基因的拷贝数)×100%。(2)血清HBsAg检测。眼眶取血每次约0.5ml,离心后血清不少于200μl。利用HBsAgCLIA试剂盒(安图生物,CL0310)检测血清中HBsAg的表达水平。HBsAg表达抑制率按如下等式计算:HBsAg表达抑制率=(1-测试组HBsAg含量/PBS对照组HBsAg含量)×100%。其中,HBsAg含量用每毫升(ml)血清含多少当量(UI)HBsAg表示。其中,对照组为本实验中施以PBS的对照组小鼠,各测试组为分别施以不同药物组合物的给药组小鼠。结果示于图2和图3中。对于单次给药后3天处理组,RBP131/X2M2和RBP131/X30M对乙肝小鼠肝组织HBV基因X基因区mRNA的抑制活性分别为92%和90%,对小鼠血清HBsAg的抑制活性分别为98%和94%;联合用药组RBP131/X2M2+X30M对乙肝小鼠肝组织HBV基因X基因区mRNA的抑制活性为97%,对小鼠血清HBsAg的抑制活性为99.3%;RBP131/NC对乙肝小鼠肝组织HBV基因X基因区mRNA及血清HBsAg均无抑制作用。对于单次给药后14天处理组,RBP131/X2M2和RBP131/X30M对乙肝小鼠肝组织HBV基因X基因区mRNA的抑制活性分别为80%和76%,对小鼠血清HBsAg的抑制活性分别为90%和78%;联合用药组RBP131/X2M2+X30M对乙肝小鼠肝组织HBV基因X基因区mRNA的抑制活性为92%,对小鼠血清HBsAg的抑制活性为95%;RBP131/NC对乙肝小鼠肝组织HBV基因X基因区mRNA及血清HBsAg均无抑制作用。上述结果证明,单独给予RBP131/X2M2和RBP131/X30M在短时间内(例如3天)均能显示出很好的抑制效果。在较长给药时间(如给药14天)后,与单独用药相比,联合用药的抑制效果显著增加。此结果说明联合用药不仅可以增强两种药物的抑制效果,而且可以使药物的持续作用时间延长。采用相同的方法,对制备实施例2中得到的RBP130/siRNA药物组合物进行了测试,测试结果与RBP131/siRNA药物组合物类似。以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1