一种交替共聚物P(OE‑alt‑CL)的制备方法及其应用与流程
本发明涉及一种交替共聚物P(OE-alt-CL)制备方法及其药物传递应用,属于聚合物合成及药物缓控释放
技术领域:
。
背景技术:
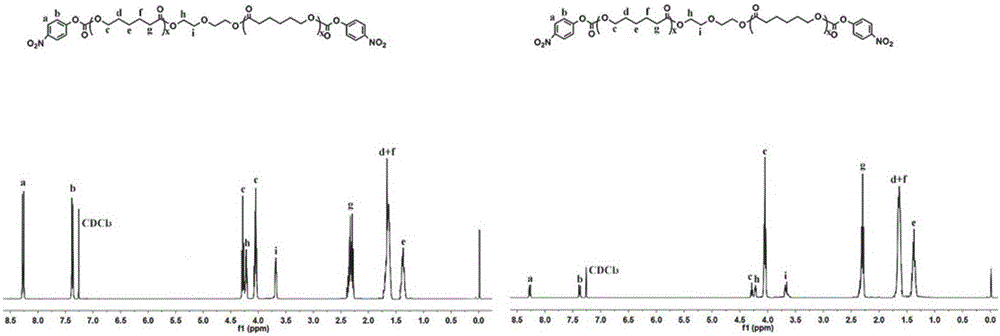
:癌症作为威胁人类健康的主要疾病之一,其发病率及致死率不断升高。治疗癌症的手段主要包括手术切除、放疗、化疗。其中化疗在癌症治疗中占据着不可替代的地位。然而,在传统的抗肿瘤药物给药方式中,小分子药物往往具有代谢快,半衰期短,血药浓度低,无肿瘤靶向性等缺点,从而使得达肿瘤部位的药物很少,因此需要多次给药来提高肿瘤部位的药物浓度,以达到治疗的效果。一般的抗肿瘤药物对于癌细胞和正常细胞的毒性没有选择性,因此传统的给药方式给患者带来了较大的毒副作用。为了解决上述缺点,纳米药物缓控释载体因其突出优点而在抗肿瘤研究中成为了热点。其优点主要包括如下几点:(1)纳米药物缓控释载体能够保护药物的活性,提高了药物的稳定性;(2)提高了药物在血液中的循环时间,这有利于药物在肿瘤部位聚集,减少给药次数,从而降低了患者的痛苦;(3)药物释放缓慢,减少了人体对药物的对抗作用,从而提高了药物的有效性和安全性;(4)通过主动靶向和被动靶向在肿瘤部位聚集。在诸多纳米药物缓控释载体中,具有环境响应性的纳米药物载体被广泛研究,其中pH响应性纳米药物载体成为了研究者的宠儿。原酸酯键相比于其他酸敏感的化学键如缩醛、缩酮、乙烯基醚等具有更高的酸敏感性,可通过调节聚原酸酯的分子量以及疏水性来调控原酸酯的降解速度,从而调节药物的释放速率,因此有望通过合理的分子设计得到一种pH响应性的缓控释药物载体。技术实现要素:一种交替共聚物P(OE-alt-CL),其结构如化学式I所示:其中OE表示原酸酯;CL表示聚己内酯,其数均分子量为500-20000;n表示整数值10-120。一种交替共聚物P(OE-alt-CL)的制备方法,具体步骤如下:将一种二氨基原酸酯单体(4,4’-二亚甲氧基-二-(2-氨基乙氧基-1,3-二氧戊烷))、聚己内酯二醇活性酯(NPC-CL-NPC),无水三乙胺和无水N,N-二甲基甲酰胺按摩尔比1:1:3:(20-50)加入到100毫升反应瓶中,通氮气常温反应3-5天后,用无水乙醚沉降3次,用乙酸乙酯洗3次,油泵负压干燥得到聚合物。一种交替共聚物P(OE-alt-CL)的制备方法,所述聚己内酯二醇活性酯(NPC-CL-NPC)的制备方法如下:将聚己内酯二醇,对硝基苯基氯甲酸酯(NPC),无水三乙胺,无水二氯甲烷按摩尔比1:3:3:(50-200)加入到250mL的圆底反应瓶中反应过夜,NaH2PO4(0.4M)萃取两次后的粗产品用硅胶柱分离得到目标产物,其中所用洗脱剂是由摩尔比为二氯甲烷:甲醇=1:2.8-3的混合溶剂组成。一种抗肿瘤纳米微球药物载体,包括上述的交替共聚物P(OE-alt-CL),以及抗肿瘤药物;其中所述的抗肿瘤药物选自喜树碱、紫杉醇、阿霉素、5-氟尿嘧啶等。一种抗肿瘤纳米微球药物载体,其制备方法如下:将交替共聚物P(OE-alt-CL),抗肿瘤药物,二氯甲烷按质量比5:1:(5-10)混合后缓慢滴加到4.8-5%聚乙烯醇水溶液中,超声后加入到0.26-0.3%聚乙烯醇水溶液中,室温搅拌3-5h,5000-15000rpm离心得到目的纳米微球药物载体,然后冻干保存。本发明的优点是:1、与传统的主链含原酸酯的聚合物的制备相比,其制备方法简单、高效、经济。2、引入了生物相容性好的疏水性聚己内酯。3、被包载在纳米微球中的药物的释放速率可控,并具有缓释的效果。附图说明附图1是实施例1中聚己内酯二醇活性酯NPC-CL-NPC的1HNMR。附图2是实施例2中交替共聚物P(OE-alt-CL)的1HNMR。附图3是实施例4中纳米微球药物载体平均粒径以及透射电镜观察的形貌,其中a,b为P(OE-alt-CL530)形成的纳米粒子NPs-1的粒径及形貌;c,d为P(OE-alt-CL2000)形成的纳米粒子NPs-2的粒径及形貌。附图4是实施例6中纳米微球药物载体包载阿霉素后的体外释放结果。附图5是实施例7中纳米微球药物载体包载阿霉素后定性检测细胞摄取结果。附图6是实施例8中纳米微球药物载体包载阿霉素后定量检测细胞摄取结果。附图7是实施例9中纳米微球药物载体包载阿霉素后的生理稳定性试验结果。附图8是实施例10中纳米微球药物载体的细胞毒性试验结果,其中a为NPs-1和NPs-2的细胞毒性;b,c为NPs-1-DOX和NPs-2-DOX的细胞毒性;d为IC50。具体实施方式实施例1聚己内酯二醇活性酯NPC-CL530-NPC的合成:向500mL的两口瓶中依次加入3.2g(6.0mmol)分子量为530的聚己内酯二醇,3.65g(18.1mmol)对硝基苯基氯甲酸酯,1.83g(18.1mmol)三乙胺,200mL的二氯甲烷,氮气环境下搅拌反应过夜,NaH2PO4(0.4M)萃取两次,粗产品用硅胶柱(洗脱剂:二氯甲烷:甲醇=1:3)进一步纯化得到油状目的产物NPC-CL530-NPC2.98g,产率57.34%。1HNMR(400MHz,CDCl3,δ,ppm):1.31-1.44(m,-CH2CH2CH2CH2CCOO-),1.56-1.73(m,-CH2CH2CH2CH2COO-),2.28-2.39(m,-CH2CH2CH2CH2COO-),3.68(t,4H,-CH2OCH2-),3.99-4.1,4.25-4.32(m,-OCOOCH2-),4.18-4.24(t,4H,-COOCH2-),7.36(d,4H,-CH=C-O-),8.26(d,4H,-CH=C-NO2)(图1)。聚己内酯二醇活性酯NPC-CL2000-NPC的合成:向500mL的两口瓶中依次加入2.4g(1.2mmol)分子量为2000的聚己内酯二醇,0.73g(3.6mmol)对硝基苯基氯甲酸酯,0.37g(3.7mmol)三乙胺,200mL的二氯甲烷,氮气环境下搅拌反应过夜,NaH2PO4(0.4M)萃取两次,粗产品用硅胶柱(洗脱剂:二氯甲烷:甲醇=1:3)进一步纯化得到油状目的产物NPC-CL2000-NPC1.38g,产率49.25%。1HNMR(400MHz,CDCl3,δ,ppm):1.31-1.44(m,-CH2CH2CH2CH2CCOO-),1.56-1.73(m,-CH2CH2CH2CH2COO-),2.28-2.39(m,-CH2CH2CH2CH2COO-),3.68(t,4H,-CH2OCH2-),3.99-4.1,4.25-4.32(m,-OCOOCH2-),4.18-4.24(t,4H,-COOCH2-),7.36(d,4H,-CH=C-O-),8.26(d,4H,-CH=C-NO2)(图1)。实施例2交替共聚物P(OE-alt-CL530)的制备:向50mL的反应瓶中依次加入1.72g(2.0mmol)聚己内酯二醇活性酯NPC-CL530-NPC,0.62g(2.0mmol)4,4’-二亚甲氧基-二-(2-氨基乙氧基-1,3-二氧戊烷),0.42g(4.1mmol)三乙胺,20mL的N,N-二甲基甲酰胺常温搅拌反应5天,用无水乙醚沉降3次,乙酸乙酯洗后,油泵负压干燥得到目的聚合物P(OE-alt-CL530)1.51g,产率87.22%。1HNMR(400MHz,CDCl3,δ,ppm):1.31-1.44(m,-CH2CH2CH2CH2COO-),1.56-1.73(m,-CH2CH2CH2CH2COO-),2.28-2.39(m,-CH2CH2CH2CH2COO-),3.37(s,-NHCH2-),3.44-3.74(m,-CH2OCH2-,-NHCH2CH2-),3.76-3.88,4.11-4.17(m,-OCH2CH-),3.99-4.11(m,-OCOOCH2-),4.18-4.24(t,-COOCH2-),4.27-4.49(m,-CH2CH(O)CH2-),5.81(d,2H,CH-C(O)3)(图2)。交替共聚物P(OE-alt-CL2000)的制备:向50mL的反应瓶中依次加入0.94g(0.4mmol)聚己内酯二醇活性酯NPC-CL2000-NPC,0.12g(0.4mmol)4,4’-二亚甲氧基-二-(2-氨基乙氧基-1,3-二氧戊烷),0.08g(0.79mmol)三乙胺,20mL的N,N-二甲基甲酰胺常温搅拌反应5天,用无水乙醚沉降3次,乙酸乙酯洗后,油泵负压干燥得到目的聚合物P(OE-alt-CL2000)0.98g,产率92.41%。1HNMR(400MHz,CDCl3,δ,ppm):1.31-1.44(m,-CH2CH2CH2CH2COO-),1.56-1.73(m,-CH2CH2CH2CH2COO-),2.28-2.39(m,-CH2CH2CH2CH2COO-),3.37(s,-NHCH2-),3.44-3.74(m,-CH2OCH2-,-NHCH2CH2-),3.76-3.88,4.11-4.17(m,-OCH2CH-),3.99-4.11(m,-OCOOCH2-),4.18-4.24(t,-COOCH2-),4.27-4.49(m,-CH2CH(O)CH2-),5.81(d,2H,CH-C(O)3)(图2)实施例3交替共聚物P(OE-alt-CL)的分子量及其分布的检测:分别取两种聚合物用DMF溶解并保证聚合物浓度为2mg/mL,用孔径为0.45μm有机相滤头过滤后,通过Waters1515GPC检测聚合物的分子量及其分布,其中以DMF为流动相,流速为1mL/min,进样量为60μL,结果如表1。实施例4纳米微球药物载体的制备将20mg的聚合物加入到0.4mL的二氯甲烷中充分溶解后,在涡旋的条件下加入到0.8mL的5%的聚乙烯醇溶液中,然后立即超声(3次,每次10s,间隔5s,功率300W),超声后立即加入到20mL的0.3%的聚乙烯醇溶液中,搅拌3h,使二氯甲烷挥发。在10000rpm下,离心10min,用0.01MpH8.0的缓冲液再分散后。通过DLS和透射电镜检测纳米载体的粒径分布及形貌,结果如图3。实施例5包载阿霉素的纳米微球药物载体的制备将20mg的聚合物和4mg的阿霉素加入到0.4mL的二氯甲烷中充分溶解后,在涡旋的条件下加入到0.8mL的5%的聚乙烯醇溶液中,然后立即超声(3次,每次10s,间隔5s,功率300W),超声后立即加入到20mL的0.3%的聚乙烯醇溶液中,搅拌3h,使二氯甲烷挥发。在10000rpm下,离心10min,用0.01MpH8.0的缓冲液再分散载药微球备用。其载药量和包封率(表2)计算公式如下:载药量(%)=纳米粒子中阿霉素的量/载药纳米粒子的总量×100%包封率(%)=纳米粒子中阿霉素的量/加入的总共阿霉素的量×100%实施例6纳米微球药物载体包载阿霉素后的体外释放检测分别取阿霉素浓度为500μg/ml的纳米粒子1mL于截留分子量为8kD-14kD的透析袋中,用棉线扎紧透析袋,放入到50mL的EP管中,再在EP管中加入5mL的缓冲液,设3个重复,在37℃,100rpm条件下振荡,分别在30min、1、2、3........12、24、36、48......120h取出缓冲液,并加入新的缓冲液,然后检测缓冲液中的阿霉素浓度,然后计算阿霉素的释放量,释放结果如图4。实施例7纳米微球药物载体包载阿霉素后定性检测细胞摄取将人神经母细胞瘤细胞(SH-SY5Y)或者小鼠肝癌细胞(H22)加入到底部带有盖玻片的12孔板中,保证每孔105个细胞左右,培养过夜后,去除培养基,加入1.8mL新鲜培养基,0.2mL的自由阿霉素以及载药纳米粒子(最终阿霉素浓度都为8μg/mL)共培养4h后,去除培养基,PBS冲洗2次,4%多聚甲醛液固定细胞(5min左右),PBS冲洗2次,染核试剂染细胞核(5min),PBS洗2次,最后用激光共聚焦显微镜观察,结果如图5。实施例8纳米微球药物载体包载阿霉素后定量检测细胞摄取将人神经母细胞瘤细胞(SH-SY5Y)或者小鼠肝癌细胞(H22)加入到6孔板中,保证每孔2.5×105个细胞左右,培养过夜后,去除培养基,加入1.8mL新鲜培养基,0.2mL的自由阿霉素以及载药纳米粒子(最终阿霉素浓度都为8μg/mL)共培养4h后,去除培养基,PBS洗2次,胰酶消化,培养基终止消化后离心收集细胞,用1mL的PBS分散后用流式细胞仪检测,结果如图6。实施例9纳米微球药物载体包载阿霉素后的生理稳定性评估将冻干载药纳米粒子分别用生理盐水,10mmolpH7.4的PBS,胎牛血清再分散,并保持浓度为1mg/mL。分别在1、3、5、7、9天用DLS检测载药纳米粒子的粒径以及光散射强度,结果如图7。实施例10纳米微球药物载体的细胞毒性检测将人神经母细胞瘤细胞(SH-SY5Y)或者小鼠肝癌细胞(H22)加入到6孔板中,保证每孔浓度在4,000个左右,贴壁24h后,去除培养基,加入180μl的新鲜培养基,20μl的自由阿霉素或者载药纳米粒子(阿霉素浓度从0.5-16μg/mL)或者空白纳米粒子(浓度从31.25-1000μg/mL),共培养24h后,去除培养基,加入180μl的新鲜培养基和20μlMTT(5mg/mL)共培养4h后,去除培养基,加入150μl的DMSO,震荡10min后,在570nm波长下检测,结果如图8。其中表1为:实施例3中交替共聚物P(OE-alt-CL)的分子量及其分子量分布。表1PolymerMn(×104)Mw(×104)PDI(Mw/Mn)P(OE-alt-CL530)2.23.811.73P(OE-alt-CL2000)4.17.871.92表2为实施例5中纳米微球药物载体包载阿霉素的载药量及包封率,其中NPs-1-DOX为NPs-1包载阿霉素后的纳米微球;NPs-2-DOX为NPs-2包载阿霉素后的纳米微球。表2SampleDLC(%)DLE(%)Diameter(nm)PDINPs-1-DOX11.456.7223.90.114NPs-2-DOX16.278.5278.70.119当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1