咖啡酸苯乙酯的晶型II及其制备方法和用途与流程
本发明涉及咖啡酸苯乙酯的一种新晶型,更具体地涉及咖啡酸苯乙酯的晶型ii及其制备方法和用途。
背景技术:
咖啡酸苯乙酯(caffeicacidphenethylester,cape)已经被鉴定为蜂胶中的主要活性组分之一,蜂胶的许多生物学活性均与cape有关,这主要表现在抗氧化、抗炎和抗肿瘤等方面。因此,cape是近年来国外蜂胶活性成分研究的一大热点。具体地,cape包括如下几方面的作用:
a)抗氧化作用
cape结构中含有儿茶酚结构,是一种强抗氧化剂。sudina实验证明,10μm的cape就能完全阻断中性粒细胞中活性氧化物质的产生,阻断黄嘌呤氧化酶系统中的活性而减少sod的消耗,从而起到抗氧化作用。
b)抗炎作用
研究表明,cape可以抑制no的产生,因此cape可以抑制nos基因的表达从而起到抗炎症的作用。cape还可以抑制前列腺素(pg)和白三烯的合成,pg是花生四烯酸(aa)在cox的催化下产生的,cox有两种异构形式分别是cox-l和cox-2,cape通过抑制细胞膜上花生四烯酸的释放,以及抑制cox-1和cox-2的活性,从而起到了抗炎作用。
c)抗肿瘤作用
蜂胶中的cape具有抗癌活性,对肿瘤细胞具有特定的杀伤力,能抑制肿瘤细胞增生。cape在极微量的浓度就可以改善由肿瘤因子(12-0-十四酰拂波醇-13-乙酸酯)引起的炎症反应。国内外研究表明,蜂胶中的cape对黑素瘤、结肠癌和胃癌细胞系的作用极为强烈,表现出抑制癌细胞的特性。
d)神经保护作用
对多种不同病因引起的神经损伤、变性及神经退行性疾病的动物模型以至临床患者显示出了明显的神经保护作用。这种对神经退行性疾病的明显治疗作用是通过针对神经损伤和退行性疾病的多个病理环节,发挥多靶点神经保护作用而产生的。该药物的作用机制包括:抑制脑和脊髓内小胶质细胞的激活,阻断神经炎症对神经元的损伤;抑制caspase-3等的活性,阻止神经元的凋亡;抑制p38丝裂原蛋白激酶的活性,阻断引起神经损伤的信号通路;抑制氧自由基的产生,阻止氧化应激对神经元的损伤;抑制inos的表达和活化,阻止no对神经元的损伤。因此,在以肌萎缩侧索硬化症(als)、帕金森氏病、老年痴呆为代表的神经退行性疾病的治疗中,cape是一具有全新机理的多靶点药物。具有极高的研究和开发价值。
神经退行性疾病(neurodegenerativedisease)是大脑和脊髓的细胞神经元丧失的疾病状态。大脑和脊髓由神经元组成,神经元有不同的功能,如控制运动,处理感觉信息,并作出决策。大脑和脊髓的细胞一般是不会再生的,所以过度的损害可能是毁灭性的,不可逆转的。神经退行性疾病是由神经元或其髓鞘的丧失所致,随着时间的推移而恶化,以导致功能障碍。神经退行性疾按表型分为两组:一类影响运动,如小脑性共济失调;一类影响记忆以及相关的痴呆症。神经退行性疾病包括阿尔兹海默病、肌肉萎缩性侧索硬化症、帕金森氏病等,目前仅有很少的药物可用于治疗该类疾病。
在配制药物组合物时,方便操作和加工的药物形态是十分重要的。不仅从获得一种商业上可行的生产方法的角度,而且从后来生产含有所述活性化合物的药用组合物的角度出发,这点都是重要的。所述活性成分的化学稳定性、固态稳定性和储存期限也是非常重要的因素。药物和包含它的组合物应该能够有效地储存超过可评估的时间周期,并且,在该周期内所述活性成分的物理-化学特征(如它的化学组成、密度、引湿性和溶解性)没有出现显著的变化。此外,能够以尽可能纯的形式提供药物也是重要的。无定形物质在这点上可能存在重大问题。例如,典型地,此类物质比结晶物质更难操作和配制,产生不可靠的溶解性,并且经常发现此类物质是不稳定的并且化学上是不纯的。技术人员将理解,如果能够获得一种以稳定晶型存在的药物,则可解决以上问题。因此,在生产商业上可行并且药学上可接受的药用组合物时,无论哪种可能,以一种充分洁净和稳定的形态提供药物都是理想的。可是,应该注意,所述目标并不总是能够实现。事实上,通常无法单从分子结构预知化合物将发生何种结晶行为,这一般是凭经验确定的。
药物多晶型现象是影响药品质量与临床疗效的重要因素之一,因此,咖啡酸苯乙酯多晶型及其制备技术对于咖啡酸苯乙酯成药而言具有非常重要的意义。因此,现有技术中还需要提供稳定性好的和/或便于制剂操作的咖啡酸苯乙酯新晶型。
技术实现要素:
本发明人在按照文献方法制备咖啡酸苯乙酯并对其研究的过程中出乎意料地发现了咖啡酸苯乙酯的新晶型(即下文所述晶型ii),所述晶型ii具有更好的稳定性,并且更利于制剂工艺过程中的操作。
本发明提供了咖啡酸苯乙酯的一种新晶型(即晶型ii),所述晶型ii的制备方法,以及所述晶型ii在生产用于预防和/或治疗神经退行性疾病的药物中的用途。本发明提供的晶型ii的制备方法具有工艺简单、收率较高且稳定性好的特点。
具体而言,本发明的技术方案如下:
本发明提供一种咖啡酸苯乙酯的晶型ii,
其中,所述咖啡酸苯乙酯具有式i所示的结构,且式i所示化合物通常命名为:(e)-3-(3,4-二羟基苯基)丙烯酸苯乙基酯;
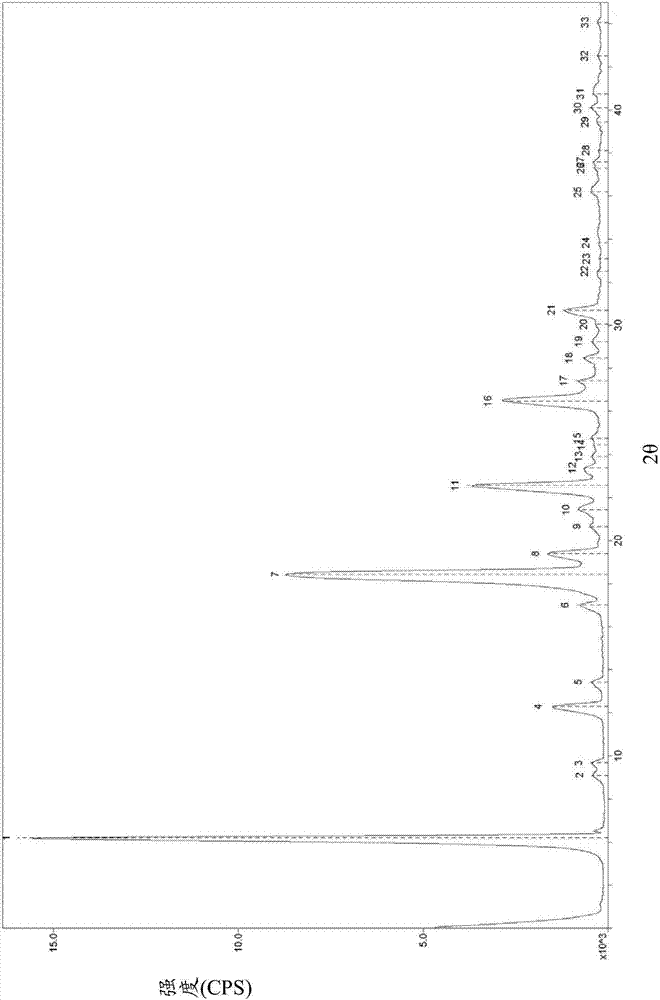
其中,所述晶型ii使用cukα辐射的x-射线粉末衍射图具有以下以2θ衍射角表示的衍射峰:6.199°、18.439°、22.560°、26.481°和30.721°;
优选地,所述晶型ii使用cukα辐射的x-射线粉末衍射图如图2所示;
优选地,所述晶型ii使用cukα辐射的x-射线粉末衍射图具有表1中以2θ衍射角及d值表示的衍射峰。
表1
还可进一步地通过dsc图谱确认和表征晶型ii,所述dsc图谱在130.7℃开始出现吸热峰,晶型ii的熔点为135.0℃,所述dsc的升温速率为10℃/min。
优选地,所述晶型ii的dsc图谱如图4所示。
另一方面,本发明提供上述咖啡酸苯乙酯的晶型ii的制备方法,所述制备方法包括以下步骤:
1)将咖啡酸苯乙酯加至溶剂a中,搅拌溶解;和
2)在搅拌下逐滴滴入一定量的溶剂b,加热使固体完全溶解,冷却,析晶,抽滤,干燥,得到晶型ii。
优选地,所述溶剂a选自c1-c4的醇、乙腈、二氧六环、丙酮、丁酮、二甲基亚砜和n,n-二甲基甲酰胺;更优选地,所述溶剂a选自异丙醇、无水乙醇和正丁醇。
优选地,所述溶剂a的使用量是咖啡酸苯乙酯的2~30倍(v/m),更优选地为4~15倍(v/m),最优选地为6~8倍(v/m);
优选地,所述溶剂b选自c4-c8的醚、c2-c8的酯、c5-c8的烷烃、甲苯和二甲苯;更优选地,所述溶剂b为甲基叔丁基醚;
优选地,所述溶剂b的使用量是咖啡酸苯乙酯的4~50倍(v/m),更优选地为10~30倍(v/m),最优选地为15~20倍(v/m);
优选地,所述加热使固体完全溶解为加热至回流使固体全部溶解;
优选地,所述冷却为冷却至室温,随后继续搅拌2-3h;
本发明的上述咖啡酸苯乙酯的晶型ii还可以通过如下步骤来制备:
i)将咖啡酸苯乙酯加至溶剂a和溶剂b的混合溶剂中,加热使固体完全溶解;和
ii)在搅拌状态下逐渐冷却,析晶,抽滤,干燥,得到晶型ii。
优选地,所述溶剂a选自c1-c4的醇、乙腈、二氧六环、丙酮、丁酮、二甲基亚砜和n,n-二甲基甲酰胺;
优选地,所述溶剂b选自c4-c8的醚、c2-c8的酯、c5-c8的烷烃、甲苯和二甲苯;
优选地,所述混合溶剂中所述溶剂a与所述溶剂b的体积比为1:0.5~1:10,更优选地为1:2~1:4;
优选地,所述混合溶剂的使用量是咖啡酸苯乙酯的6~80倍(v/m),更优选地为14~45倍(v/m),最优选地为21~28倍(v/m);
优选地,所述加热使固体完全溶解为加热至回流温度下搅拌0.5-1h使固体全部溶解;
优选地,所述冷却为冷却至室温,随后继续搅拌2-3h;
又一方面,本发明提供上述咖啡酸苯乙酯晶型ii在制备用于预防和/或治疗神经退行性疾病的药物中的用途。
优选地,所述神经退行性疾病选自阿尔兹海默病、肌肉萎缩性侧索硬化症和帕金森氏病等。
事实上,在研究本发明的咖啡酸苯乙酯晶型的过程中,本发明人还发现咖啡酸苯乙酯的晶型i的形态,但晶型i没有前述的晶型ii稳定,为了对比作用,以下对于咖啡酸苯乙酯晶型i及其制备方法进行了描述。
咖啡酸苯乙酯的晶型i使用cukα辐射的x-射线粉末衍射图具有以下以2θ衍射角表示的衍射峰:5.241°、6.220°、16.179°、17.920°、18.439°、20.181°、22.300°、24.839°、26.580°;
优选地,咖啡酸苯乙酯的晶型i使用cukα辐射的x-射线粉末衍射图如图1所示。
在咖啡酸苯乙酯的晶型i的dsc分析图中,其在129℃开始出现吸热峰,晶型i的熔点为134.3℃,所述dsc的升温速率为10℃/min。
优选地,所述晶型i的dsc图谱如图3所示。
本发明还公开了咖啡酸苯乙酯晶型i的制备方法,但不仅限于下列方法:
将咖啡酸苯乙酯加至特定溶剂中,搅拌溶解,在搅拌下滴入5~15倍(v/m)咖啡酸苯乙酯的纯化水,继续搅拌直至析出晶体。其中所述的溶剂选自c1-c4的醇、乙腈、二氧六环、丙酮、丁酮、二甲基亚砜和n,n-二甲基甲酰胺。
由于多晶型批次之间以及与非晶形态之间的不同物理性质明显影响化合物的化学和药用加工。特别是当以工业规模制备或使用所述化合物时尤为如此。因此,提供稳定性好的和/或便于制剂操作的晶型显得尤为重要。针对这一技术难题,本发明提供了一种咖啡酸苯乙酯的新晶型及其制备方法和用途,采用本发明的方法制备的晶型ii纯度较高且稳定性好,并且本发明的方法简便可靠,可重复性好,环境友好性佳。
本发明公开的咖啡酸苯乙酯晶型i和晶型ii可通过参考它们的差示扫描量热法(dsc)的热分析图和粉末x-射线衍射图进行区别。通常,对于同种化合物的同种晶型,其x射线衍射图在整体上具有相似性,表征峰位置的d值误差一般在±2%之内,大部分误差不会超过±2%,相对强度误差可能较大,但变化趋势一致。另外,在混合物的鉴定中,由于含量下降等因素会造成部分衍射线缺失,此时无需依赖高纯试样中观察到的全部谱带,甚至一条谱带也可能对给定的结晶是特征性的。确定本发明的说明书及权利要求书中的粉末x射线衍射图谱的衍射角2θ时,所得的值应理解为在该值的±1.0°的范围内,较好是在该值±0.2°的范围内。
附图说明
以下,结合附图来详细说明本发明的实施方案,其中:
图1为咖啡酸苯乙酯晶型i的粉末x-射线衍射图,其中纵轴表示峰强度,横轴表示衍射角(2θ)。
图2为咖啡酸苯乙酯晶型ii的粉末x-射线衍射图,其中纵轴表示峰强度,横轴表示衍射角(2θ)。
图3为咖啡酸苯乙酯晶型i的dsc热分析图,其中纵轴表示mw/mg,即单位质量的样品随温度变化所吸收或放出的能力,横轴表示温度℃。
图4为咖啡酸苯乙酯晶型ii的dsc热分析图,其中纵轴表示mw/mg,即单位质量的样品随温度变化所吸收或放出的能力,横轴表示温度℃。
具体实施方式
以下实施例进一步描述本发明,但是,这些实施例仅是用于说明本发明,而不是对本发明范围的限制。
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的原料、试剂等,如无特殊说明,均为市售购买产品。
本发明中图谱测定所用的仪器和测定条件如下:
dsc:
仪器型号:微机差热天平hct-1,北京恒久科学仪器厂生产。
测试条件与方法:测试温度范围为室温至600℃,升温速率为10℃/min,载气为氮气,tga-dsc联用测定其热分析图谱。
仪器的校正与检定:按照中国药品检验标准操作规范2010年版的方法,以标准物质铟对热分析仪进行校准。
xrd:
仪器型号:全自动多晶x射线衍射仪dmax/2400。
测试条件与方法:照x-射线粉末衍射法(中国药典2010年版二部附录ⅸf)测定,参数设定为石墨单色器,cukα,扫描范围2θ为3°-60°,扫描速度为4°/min,步长为0.02°,管压管流为36kv/20ma。
实施例1咖啡酸苯乙酯的制备
咖啡酸苯乙酯为北京红惠新医药科技有限公司自制,其化学方程式及制备方法如下:
将1l甲苯加入到3l反应瓶中,机械搅拌下依次加入sm1(丙二酸环(亚)异丙酯,240克,市售)和sm2(苯乙醇,202克,市售),加料完毕后,升温至100℃开始计时,8小时后停止反应,自然冷却降温至室温,继续搅拌的同时加入sm3(3,4-二羟基苯甲醛,160克,市售),然后依次加入吡啶166ml和哌啶17ml,随后升温至内温到95℃,反应15个小时后停止反应,降温至室温,加入1l乙酸乙酯,随后依次用1l1m盐酸溶液和1l饱和食盐水洗涤有机相,分液,有机相用无水硫酸钠干燥30分钟后,过滤,真空旋蒸掉有机溶剂得粗品,粗品在2l正己烷里打浆后过滤,滤饼真空干燥箱内40℃24小时后得到270克咖啡酸苯乙酯产品。
实施例2咖啡酸苯乙酯晶型i的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),10ml无水乙醇,搅拌溶解。保持搅拌状态,向体系中滴入20ml纯化水,搅拌析晶。所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.6g咖啡酸苯乙酯i型晶体。
分别以x射线粉末衍射法和差示扫描量热法表征获得的晶体,结果示于图1和图3。
实施例3咖啡酸苯乙酯晶型i的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),8ml丙酮,搅拌溶解。保持搅拌状态,向体系中滴入15ml纯化水,搅拌析晶。所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.7g咖啡酸苯乙酯i型晶体。其x射线粉末衍射图和dsc热分析图分别与图1和图3相似。
实施例4咖啡酸苯乙酯晶型i的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),12ml甲醇,搅拌溶解。保持搅拌状态,向体系中滴入18ml纯化水,搅拌析晶。所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.4g咖啡酸苯乙酯i型晶体。其x射线粉末衍射图和dsc热分析图分别与图1和图3相似。
实施例5咖啡酸苯乙酯晶型ii的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),14ml无水乙醇,搅拌溶解。保持搅拌状态,向体系中滴入30ml甲基叔丁基醚,加热回流搅拌全溶后,慢慢降温析晶,降至室温后继续搅拌2小时,随后将所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.6g咖啡酸苯乙酯ii型晶体。
分别以x射线粉末衍射法和差示扫描量热法表征获得的晶体,结果示于图2和图4。
实施例6咖啡酸苯乙酯晶型ii的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),14ml异丙醇,搅拌溶解。保持搅拌状态,向体系中滴入30ml甲基叔丁基醚,加热回流搅拌全溶后,慢慢降温析晶,降至室温后继续搅拌2.5小时,随后将所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.75g咖啡酸苯乙酯ii型晶体。
其x射线粉末衍射图和dsc热分析图分别与图2和图4相似。
实施例7咖啡酸苯乙酯晶型ii的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),5ml丙酮和20ml甲苯,加装回流冷凝装置。保持搅拌状态,升温至回流。保持回流1小时确保固体完全溶解。关闭加热,保持搅拌缓慢降温析晶。所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.45g咖啡酸苯乙酯ii型晶体。其x射线粉末衍射图和dsc热分析图分别与图2和图4相似。
实施例8咖啡酸苯乙酯晶型ii的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),12ml乙腈和25ml甲基叔丁基醚,加装回流冷凝装置。保持搅拌状态,升温至回流。保持回流1小时确保固体完全溶解。关闭加热,保持搅拌缓慢降温析晶。所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.5g咖啡酸苯乙酯ii型晶体。其x射线粉末衍射图和dsc热分析图分别与图2和图4相似。
实施例9咖啡酸苯乙酯晶型ii的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),15ml二氧六环和40ml正己烷,加装回流冷凝装置。保持搅拌状态,升温至回流。保持回流1小时确保固体完全溶解。关闭加热,保持搅拌缓慢降至室温后继续搅拌3小时,。所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.6g咖啡酸苯乙酯ii型晶体,本结晶体系结晶收率较高。其x射线粉末衍射图和dsc热分析图分别与图2和图4相似。
实施例10咖啡酸苯乙酯晶型ii的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),16ml丁酮和35ml甲苯,加装回流冷凝装置。保持搅拌状态,升温至回流。保持回流1小时确保固体完全溶解。关闭加热,保持搅拌缓慢降温析晶。所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.4g咖啡酸苯乙酯ii型晶体。其x射线粉末衍射图和dsc热分析图分别与图2和图4相似。
实施例11咖啡酸苯乙酯晶型ii的制备
于100ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),3mln,n-二甲基甲酰胺和3ml乙酸乙酯及15ml甲苯,加装回流冷凝装置。保持搅拌状态,升温至回流。保持回流1小时确保固体完全溶解。关闭加热,保持搅拌缓慢降温析晶,降至室温后继续搅拌2小时,随后将所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得1.2g咖啡酸苯乙酯ii型晶体。其x射线粉末衍射图和dsc热分析图分别与图2和图4相似。
实施例12咖啡酸苯乙酯晶型ii的制备
于100ml~150ml单口烧瓶中加入2g咖啡酸苯乙酯(由实施例1制备),xml溶剂a,搅拌溶解。保持搅拌状态,向体系中滴入yml溶剂b,加热回流搅拌全溶后,慢慢降温析晶,降至室温后继续搅拌2.5小时,随后将所得悬浮液抽滤,滤饼在45℃真空干燥6h,即得zg咖啡酸苯乙酯ii型晶体。分别以x射线粉末衍射法和差示扫描量热法表征获得的晶体,其中,除试验号为14和19的实验未获得晶体外,其它实验获得的晶体的表征结果分别与图2和图4相似。其中溶剂a和溶剂b的种类及用量汇总于表2:
表2
实施例13咖啡酸苯乙酯晶型i和晶型ii的稳定性研究
对咖啡酸苯乙酯晶型i(由实施例1制备)和晶型ii(由实施例5制备)分别进行了稳定性研究:影响因素实验(高温60℃、高湿90%±5%、光照4500lx)、加速试验(温度40±2℃、相对湿度75%±5%)和长期试验(温度25±2℃、相对湿度60%±5%)及经研磨、压片后对样品进行x射线粉末衍射测试,试验结果如表3所示:
表3
试验结果表明:本品经研磨、压片后,经x-射线粉末衍射测试,主要2θ角均未发生显著变化,说明本品在制剂过程中晶型稳定性较好。晶型i在加速试验6个月和长期试验12个月后,晶型发生一定变化;但晶型ii在上述加速试验和长期试验后晶型未发生明显变化,说明本品晶型ii在存储过程中晶型稳定,符合药用需求。
- 还没有人留言评论。精彩留言会获得点赞!